
An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Elsevier - PMC COVID-19 Collection

Membrane Transport
Life depends on a membrane's ability to precisely control the level of solutes in the aqueous compartments, inside and outside, bathing the membrane. The membrane determines what solutes enter and leave a cell. Transmembrane transport is controlled by complex interactions between membrane lipids, proteins, and carbohydrates. How the membrane accomplishes these tasks is the topic of this chapter.
1. Introduction
Life depends on a membrane's ability to precisely control the level of solutes in the aqueous compartments, inside and outside, bathing the membrane. The membrane determines what solutes enter and leave a cell. Transmembrane transport is controlled by complex interactions between membrane lipids, proteins, and carbohydrates. How the membrane accomplishes these tasks is the topic of Chapter 19.
A biological membrane is semipermeable, meaning it is permeable to some molecules, most notably water, while being very impermeable to most solutes (various biochemicals and salts) found in the bathing solution. This very important concept of unequal transmembrane distribution and, hence, permeability between water and other solutes came out of the pioneering work of Charles Overton in the 1890s (see Chapter 2). How does a biological membrane accomplish semipermeability? The barrier to solute movement is largely provided by the membrane's hydrophobic core, a very thin (∼40 Å thick), oily layer. The inherent permeability of this core varies from membrane to membrane. Generally, the more tightly packed the lipids comprising the bilayer, the lower its permeability will be. Lipid bilayers are very impermeable to most solutes because of their tight packing. Fig. 19.1 depicts the membrane permeability of a variety of common solutes [1] . Note the data are presented as a log scale of solute permeability ( P in cm/s) and ranges from Na + = 10 −12 cm/s to water = 0.2 × 10 −2 cm/s, spanning almost 10 orders of magnitude!

Log of the permeability ( P in cm/s) across lipid bilayer membranes for common solutes ranging from Na + (10 −12 cm/s) to water (0.2 × 10 −2 cm/s). This range spans almost 10 orders of magnitude [1] .
Lipid bilayer permeability is not a constant but instead is affected by environmental factors. For example, LUVs (large unilamellar veicles) made from DPPC (16:0, 16:0 PC) have a sharp phase transition temperature, T m , of 41.3°C. At temperatures well below T m , the LUVs are in the tightly packed gel state and permeability is extremely low. At temperatures well above T m , the LUVs are in the loosely packed liquid disordered state ( l d , also called the liquid crystalline state) and permeability is high. However, maximum permeability is not found in the l d state, but rather at the T m [2] . As the LUVs are heated from the gel state and approach the T m , domains of l d start to form in the gel state. Solutes can then pass more readily through the newly formed l d domains than the gel domains resulting in an increase in permeability. At T m there is a maximum amount of coexisting gel and l d state domains that exhibit extremely porous domain boundaries. It is through these boundaries that most permeability occurs. As the temperature is further increased, the LUVs pass into the l d state and the interface boundaries disappear, reducing permeability to that observed for the single-component l d state. Thus, maximum permeability is observed at the T m .
1.1. Fick's First Law
The tendency for solutes to move from a region of higher concentration to one of lower concentration was first defined in 1855 by the physiologist Adolf Fick ( Fig. 19.2 ). His work is summarized in what is now the very well-known Fick's Laws of Diffusion [3] . The laws apply to both free solution and diffusion across membranes. Fick developed his laws by measuring concentrations and fluxes of salt diffusing between two reservoirs through connecting tubes of water.

Adolf Fick, 1829–1901.
Fick's First Law describes diffusion as:
Where D = diffusion coefficient (bigger molecules have lower D s); A = cross-sectional area over which diffusion occurs; dc / dx is the solute concentration gradient (diffusion occurs from a region of higher concentration to one of lower concentration).
The relationship between a solute's molecular weight and its diffusion coefficient is shown in Table 19.1 . Large solutes have low diffusion coefficients and therefore diffuse more slowly than small solutes. The diffusion rate for a particular solute under physiological conditions is a constant and cannot be increased. This defines the theoretical limit for an enzymatic reaction rate and also limits the size of a cell. If a solute starts at the center of a bacterial cell, it takes about 10 −3 s to diffuse to the plasma membrane. For this reason, typical cells are microscopic (see Chapter 1). At about 3.3 pounds and the size of a cantaloupe, the largest cell on Earth today is the ostrich egg. However a fossilized dinosaur egg in the American Museum of Natural History in New York is about the size of basketball. Since an egg's only function is to store nutrients for a developing embryo, its size is many orders of magnitude larger than a normal cell.
Table 19.1
Relationship Between a Solute's Molecular Weight and Its Diffusion Coefficient, D
| Compound | O | Acetyl choline | Sucrose | Serum albumin |
|---|---|---|---|---|
| (cm /s × 10 ) | 19.8 | 5.6 | 2.4 | 0.7 |
| Molecular weight | 32 | 182 | 342 | 69,000 |
1.2. Osmosis
Osmosis is a special type of diffusion, namely the diffusion of water across a semipermeable membrane. Water readily crosses a membrane down its potential gradient from high to low potential ( Fig. 19.3 ) [4] . Osmotic pressure is the force required to prevent water movement across the semipermeable membrane. Net water movement continues until its potential reaches zero. An early application of the basic principles of osmosis came from the pioneering work on hemolysis of red blood cells by William Hewson in the 1770s (see Chapter 2). It has also been discussed that MLVs (multilamellar vesicles, liposomes) behave as almost perfect osmometers, swelling in hypotonic solutions and shrinking in hypertonic solutions (see Chapter 3) [5] , [6] . Liposome swelling and shrinking can be easily followed by changes in absorbance due to light scattering using a simple spectrophotometer. Therefore, osmosis has been investigated for many years using common and inexpensive methodologies and a lot is known about the process.

Osmosis and osmotic pressure. Water is placed in a U-shaped tube where each of the tube arms is separated by a semipermeable membrane with pores of a size that water can easily pass through but a solute cannot. Upon addition of the solute to the tube's right arm, water diffuses from left to right (high water potential to low). The column of water in the tube's right arm (the one containing the solute) rises until the extra weight of the column equals the osmotic pressure caused by the solute. A pump could then be used to counter the osmotic pressure whereupon the solution columns in the right and left arms of the tube are made the same. The pump pressure required to equalize the height of the two columns is the osmotic pressure [4] . Note a small amount of the solute leaks from right to left since no filter is perfect.
Membranes are rarely, if ever, perfectly semipermeable. Deviation from ideality is defined by a reflection coefficient ( σ ). For an ideal semipermeable membrane where a solute is totally impermeable, σ = 1. If a solute is totally permeable (its permeability is equal to water), σ = 0. Biological membranes are excellent semipermeable barriers with σ = 0.75 to 1.0.
2. Simple Passive Diffusion
Movement of solutes across membranes can be divided into two basic types: passive diffusion and active transport [7] . Passive diffusion requires no additional energy source other than what is found in the solute's electrochemical (concentration) gradient and results in the solute reaching equilibrium across the membrane. Passive diffusion can be either simple passive diffusion where the solute crosses the membrane anywhere by simply dissolving into and diffusing through the lipid bilayer, or facilitated passive diffusion where the solute crosses the membrane at specific locations where diffusion is assisted by solute-specific facilitators or carriers. Active transport requires additional energy, often in the form of ATP, and results in a nonequilibrium, net accumulation (uptake) of the solute on one side of the membrane. The basic types of membrane transport, simple passive diffusion, facilitated diffusion (by channels and carriers) and active transport are summarized in Fig. 19.4 [8] . There are countless different examples of each type of membrane transport process [7] . Only a few representative examples will be discussed here.

Basic types of membrane transport, simple passive diffusion, facilitated diffusion (by channels and carriers), and active transport [8] .
Even simple passive diffusion requires energy to cross a bilayer membrane. In order to cross a membrane, the solute must first lose its waters of hydration, diffuse across the membrane, and then regain its waters on the opposite side. The limiting step involves the energy required to lose the waters of hydration. Table 19.2 shows the relationship between the waters of hydration (proportional to the number of —OH groups on a homologous series of solutes) and the activation energy for transmembrane diffusion. As the number of waters of hydration increases from glycol < glycerol < erythritol, the activation energy for diffusion also increases. The activation energy compares very well with the energy of hydration.
Table 19.2
Relationship Between the Waters of Hydration (Number of —OH Groups on a Homologous Series of Solutes) and the Activation Energy for Transmembrane Diffusion
| Solute | Activation energy (kJ/mol) |
|---|---|
| Glycol (HO—CH —CH —OH) | 60 |
| Glycerol (HO—CH —CH(OH)—CH —OH) | 77 |
| Erythritol (HO—CH —CH(OH)—CH(OH)—CH —OH) | 87 |
However, water diffusion does not fit this model. Water permeability is just too high. Several possibilities have been suggested to account for the abnormally high membrane permeability of water:
- 1. Water is very small and so it just dissolves in bilayers better than larger solutes.
- 2. Due to its size, water can readily enter very small statistical pores (∼4.2 Å in diameter). Statistical pores result from the simultaneous lateral movement of adjacent membrane phospholipids in opposite directions. Statistical pores have only a fleeting existence and cannot be isolated or imaged.
- 3. Passage down water chains.
- 4. Water can be carried down kinks in acyl chains that result from acyl chain melting (see lipid melting in Chapter 9).
- 5. Water may rapidly cross membranes through nonlamellar regions (eg, micelles, cubic or H II phase—see Chapter 10).
- 6. High water permeability will occur at regions of packing defect (eg, surface of integral membrane proteins, boundary between membrane domains).
- 7. Through pores or channels used to conduct ions.
- 8. Through specific water channels known as aquaporins (see below, Chapter 19, Section 3.5 ).
The only molecules that can cross a membrane by simple passive diffusion are water, small noncharged solutes, and gasses. Charged or large solutes are virtually excluded from membranes and so require more than just simple passive diffusion to cross a membrane.
3. Facilitated Diffusion
Facilitated diffusion (also known as carrier-mediated diffusion) is, like simple passive diffusion, dependent on the inherent energy in a solute gradient. No additional energy is required to transport the solute and the final solute distribution reaches equilibrium across the membrane. Facilitated diffusion, unlike simple passive diffusion, requires a highly specific transmembrane integral protein or carrier to assist in the solute's membrane passage. Facilitators come in two basic types: carriers and gated channels. Facilitated diffusion exhibits Michaelis-Menton saturation kinetics ( Fig. 19.5 , Part A, right), indicating the carrier has an enzyme-like active site. Like enzymes, facilitated diffusion carriers exhibit saturation kinetics and recognize their solute with exquisite precision, easily distinguishing chemically similar isomers like d -glucose from l -glucose. Fig. 19.5 (Part A) compares simple passive diffusion to facilitated diffusion. The figure is not to scale, however, as facilitated diffusion is orders of magnitude faster than simple passive diffusion.

(A) Simple passive diffusion (top, left) and facilitated passive diffusion (top, right) both result in a final equilibrium distribution of a solute across the membrane. For a noncharged solute, the final distribution of the solute would find equal amounts of S on both sides of the membrane. Facilitated diffusion employs a specific transporter and exhibits Michaelis–Menten saturation kinetics. (A, center right) Active transport (bottom) utilizes energy, often in the form of ATP, to drive solute uptake against its gradient resulting in a net accumulation of the solute.
3.1. Glucose Transporter
A well-studied example of a facilitated diffusion carrier is the glucose transporter, or GLUT [9] . From the activation energies for transmembrane simple passive diffusion of glycol, glycerol and erythritol presented in Table 19.2 , it can be estimated that the activation energy for glucose should be well over 100 kJ/mol, but instead it is only 16 kJ/mol. This large discrepancy is attributed to the presence of a glucose-facilitated diffusion carrier. Fig. 19.6 demonstrates the mode of action of one of these transporters, GLUT-1, from the erythrocyte [10] . GLUTs occur in nearly all cells and are particularly abundant in cells lining the small intestine. GLUTs are but one example in a superfamily of transport facilitators. GLUTs are integral membrane proteins whose membrane-spanning region is composed of 12 α-helices. GLUTs function through a typical membrane transport mechanism [10] . Glucose binds to the membrane outer surface site causing a conformational change associated with transport across the membrane. At the inner side of the membrane, glucose is released into the internal aqueous solution ( Fig. 19.6 ).

Glucose-facilitated diffusion transporter GLUT-1 [10] .
3.2. Potassium Channels
In virtually all organisms there exists a wide variety of ion channels, the most widely distributed being potassium channels [11] . There are four basic classes of potassium channels, all of which provide essential membrane-associated functions including setting and shaping action potentials and hormone secretion:
- 1. Calcium-activated potassium channel
- 2. Inwardly rectifying potassium channel
- 3. Tandem pore domain potassium channel
- 4. Voltage-gated potassium channel
Potassium channels are composed of four protein subunits that can be the same (homotetramer) or closely related (heterotetramer). All potassium channel subunits have a distinctive pore-loop structure that sits at the top of the channel and is responsible for potassium selectivity [12] . This is often referred to as a selectivity or filter loop. The selectivity filter strips the waters of hydration from the potassium ion, allowing it into the channel. Farther down the structure is a 10-Å-diameter, transmembrane, water-filled central channel that conducts potassium across the membrane. Elucidating the three-dimensional structure of this important integral membrane protein by X-ray crystallography ( Fig. 19.7 ) [12] was a seminal accomplishment in the field of membrane biophysics. For this work from 1998, Rod MacKinnon ( Fig. 19.8 ) of Rockefeller University was awarded the 2003 Nobel Prize in Chemistry. Until the potassium channel work, just obtaining the structure of non–water-soluble proteins was next to impossible. MacKinnon's work elucidated not only the structure of the potassium channel but also its molecular mechanism. It has served as a blueprint for determining the structure of other membrane proteins and has greatly stimulated interest in the field.

Three-dimensional structure of the potassium channel [12] . The channel itself is the clear opening in the center of the structure and a single K + is shown in the center of the channel.

Rod MacKinnon, 1956–.
3.3. Sodium Channel
In some ways, Na + channels [13] parallel the action of K + channels. They are both facilitated diffusion carriers that conduct the cation down the ion's electrochemical gradient. In excitable cells such as neurons, myocytes, and some glia, Na + channels are responsible for the rising phase of action potentials (see Chapter 18). Therefore agents that block Na + channels also block nerve conduction and so are deadly neurotoxins. There are two basic types of Na + channels: voltage-gated and ligand-gated. The opening of a Na + channel has a selectivity filter that attracts Na + . From there the Na + ions flow into a constricted part of the channel that is about 3–5 Å wide. This is just large enough to allow the passage of a single Na + with one attached water. Since the larger K + cannot squeeze through, the channel is selective for Na + . Of particular interest are two extremely potent biological toxins, tetrodotoxin (TTX) and saxitoxin (STX) ( Fig. 19.9 , [14] ), that, in seafood, have killed and injured many humans. Both toxins shut down Na + channels by binding from the extracellular side.

Structures of the extremely potent neurotoxins, tetrodotoxin (TTX) and saxitoxin (STX). Both neurotoxins function by blocking the Na + channel.
TTX is encountered primarily in puffer fish but also in porcupine fish, ocean sunfish, and triggerfish. TTX ( Fig. 19.9 , left) is a potent neurotoxin that blocks Na + channels while having no effect on K + channels. Puffer fish is the second most poisonous vertebrate in the world, trailing only the Golden Poison Frog that is endemic to the rain forests on the Pacific Coast of Colombia. In some parts of the world puffer fish are considered to be a delicacy but must be prepared by chefs who really know their business, as a slight error can be fatal. Puffer poisoning usually results from consumption of incorrectly prepared puffer soup, and TTX has no known antidote!
Saxitoxin (STX, Fig. 19.9 , right) is a Na + channel–blocking neurotoxin produced by some marine dinoflagellates that can accumulate in shellfish during toxic algal blooms known as Red Tide. Saxitoxin is one of the most potent natural toxins, and it has been estimated that a single contaminated mussel has enough STX to kill 50 humans! STX's toxicity has not escaped the keen eye of the United States military, which has weaponized the toxin and given it the designation TZ.
3.4. Solute Equilibrium
The driving force for transmembrane solute movement by simple or passive diffusion is determined by the free energy change, Δ G .
Where Δ G is the free energy change; [ s o ′ ] is the solute concentration on the right side of a membrane; [ s o ] is the solute concentration on the left side of a membrane; R is the gas constant; T is the temperature in K; Z is the charge of the solute; F is the Faraday; ΔΨ is the transmembrane electrical potential.
Solute movement will continue until Δ G = 0. If Δ G is negative, solute movement is left to right (it is favorable as drawn). If Δ G is positive, solute movement is right to left (it is unfavorable in the left-to-right direction) or energy must be added for the solute to go from left to right. The equation has two parts; a transmembrane chemical gradient ( [ s o ′ ] / [ s o ] ) and a transmembrane electrical gradient (ΔΨ). The net movement of a solute is therefore determined by a combination of the solute's chemical gradient and an electrical gradient inherent to the cell. If the solute has no charge, Z = 0 (as is the case for glucose) and the right hand part of the equation ( Z FΔΨ) drops out. Therefore, the final equilibrium distribution of glucose across the membrane will have the internal glucose concentration equal to the external glucose concentration and is independent of ΔΨ, the electrical potential. At equilibrium for a noncharged solute, Δ G = R T ln [ s o ′ ] / [ s o ] and Δ G can only be = zero if [ s o ′ ] = [ s o ] .
The situation for a charged solute like K + is more complicated. The net Δ G is determined by both the chemical gradient ( [ s o ′ ] / [ s o ] ) and electrical gradient (ΔΨ). The ΔΨ results from the sum of all charged solutes on both sides of the membrane, not just K + . Therefore even if the K + concentration is higher inside the cell than outside (the chemical gradient is unfavorable for K + uptake), the ΔΨ may be in the correct direction (negative interior) and of sufficient magnitude to drive K + uptake against its chemical gradient.
3.5. Aquaporins
Aquaporins are also known as water channels and are considered to be “the plumbing system for cells” [15] , [16] . For decades it was assumed that water simply leaked through biological membranes by numerous processes described above (Chapter 19, Section 2 ). However, these methods of water permeability could not come close to explaining the rapid movement of water across some cells. Although it had been predicted that water pores must exist in very leaky cells, it was not until 1992 that Peter Agre ( Fig. 19.10 ) at Johns Hopkins University identified a specific transmembrane water pore that was later called aquaporin-1. For this accomplishment Agre shared the 2003 Nobel Prize in Chemistry with Rod MacKinnon for his work on the potassium channel. Aquaporins are usually specific for water permeability and exclude the passage of other solutes. A type of aquaporin known as aqua-glyceroporins can also conduct some very small uncharged solutes such as glycerol, CO 2 , ammonia, and urea across the membrane. However, all aquaporins are impermeable to charged solutes. Water molecules traverse the aquaporin channel in single file ( Fig. 19.11 ) [17] .

Peter Agre, 1949–.

Aquaporin. Water molecules pass through the aquaporin channel in single file.
4. Active Transport
A characteristic of all living membranes is the formation and maintenance of transmembrane gradients of all solutes including salts, biochemicals, macromolecules, and even water. In living cells, large gradients of Na + and K + are particularly important. Typical cell concentrations are:
| Cell interior: | 400 mmol/L K , 50 mmol/L Na |
| Cell exterior: | 20 mmol/L K , 440 mmol/L Na |
Living cells will also have a ΔΨ from −30 to −200 mV (negative interior) resulting from the uneven distribution of all ionic solutes including Na + and K + . The chemical and electrical gradients are maintained far from equilibrium by a multitude of active transport systems. Active transport requires a form of energy (often ATP) to drive the movement of solutes against their electrochemical gradient, resulting in a nonequilibrium distribution of the solute across the membrane. A number of nonexclusive and overlapping terms are commonly used to describe the different types of active transport. Some of these are depicted in Fig. 19.12 [18] .

Basic types of active transport [18] .
4.1. Primary Active Transport
Primary active transport is also called direct active transport or uniport. It involves using energy (usually ATP) to directly pump a solute across a membrane against its electrochemical gradient.
The most studied example of primary active transport is the plasma membrane Na + ,K + -ATPase discussed below (Chapter 19, Section 4.2 ). Other familiar examples of primary active transport are the redox H + -gradient generating system of mitochondria (see Chapter 18), the light-driven H + -gradient generating system of photosynthetic thylakoid membranes, and the ATP-driven acid (H + ) pump found in the epithelial lining of the stomach. There are four basic types of ATP-utilizing primary active transport systems ( Table 19.3 ).
Table 19.3
Four Types of ATP-Using Primary Active Transport Systems
| ATP-using primary active transport systems | Example |
|---|---|
| P-type | Na ,K -ATPase Ca pump H acid pump |
| F-type | Mitochondrial ATP synthase Chloroplast ATP synthase |
| V-type | Vacuolar ATPase |
| ABC (ATP binding cassette transporter) | Many |
4.2. Na + ,K + -ATPase
Arguably the most important active transport protein is the plasma membrane-bound Na + ,K + -ATPase. This single enzyme accounts for one-third of human energy expenditure and is often referred to as the “pacemaker for metabolism.” As a result the Na + ,K + -ATPase has been extensively studied for more than 50 years. The enzyme was discovered in 1957 by Jens Skou ( Fig. 19.13 ) who, 40 years later, was awarded the 1997 Nobel Prize in Chemistry.

Jens Skou, 1918–.
As is often the case in biochemistry, a serendipitous discovery of a natural product from the jungles of Africa has been instrumental in unraveling the enzyme's mechanism of action. The compound is ouabain ( Fig. 19.14 ), a cardiac glycoside first discovered in a poison added to the tip of Somali tribesmen's hunting arrows. In fact the name ouabain comes from the Somali word waabaayo that means “arrow poison.” The sources of ouabain are ripe seeds and bark of certain African plants and ouabain is potent enough to kill a hippopotamus with a single arrow. For decades after its discovery, ouabain was routinely used to treat atrial fibrillation and congestive heart failure in humans. More recently, ouabain has been replaced by digoxin, a structurally related, but more lipophilic cardiac glycoside.

Structure of ouabain.
There are several important observations about Na + ,K + -ATPase that had to be factored in before a mechanism of action could be proposed. These include:
- 1. Na + ,K + -ATPase is an example of active antiport and primary active transport.
- 2. Na + ,K + -ATPase is inhibited by ouabain, a cardiac glycoside.
- 3. Ouabain binds to the outer surface of Na + ,K + -ATPase and blocks K + transport into the cell.
- 4. Na + binds better from the inside.
- 5. K + binds better from the outside.
- 6. ATP phosphorylates an aspartic acid on the enzyme from the inside.
- 7. Phosphorylation is related to Na + transport.
- 8. Dephosphorylation is related to K + transport.
- 9. Dephosphorylation is inhibited by ouabain.
- 10. Three Na + ions are pumped out of the cell as two K + ions are pumped in, driven by hydrolysis of one ATP.
- 11. Na + ,K + -ATPase is electrogenic.
Mechanism of Na + ,K + -ATPase [19] is based on toggling back and forth between two conformational states of the enzyme, ENZ-1 and ENZ-2 ( Fig. 19.15 ). Three Na + s bind from the inside to Na + ,K + -ATPase in one conformation (ENZ-1). This becomes phosphorylated by ATP causing a conformation change producing ENZ-2∼P. ENZ-2∼P does not bind Na + , but does bind two K + ions. Therefore, three Na + ions are released to the outside and two K + ions are bound from the outside, generating ENZ-2∼P (2K + ). Upon hydrolysis of ∼P, Na + ,K + -ATPase (ENZ II) reverts back to the original ENZ-1 conformation that releases two K + ions and binds three Na + ions from the inside. Ouabain blocks the dephosphorylation step.

Mechanism of the Na + ,K + -ATPase.
4.3. Secondary Active Transport
Secondary active transport (also known as cotransport) systems are composed of two separate functions. The energy-dependent movement of an ion (eg, H + , Na + , or K + ) generates an electrochemical gradient of the ion across the membrane. This ion gradient is coupled to the movement of a solute in either the same direction (symport) or in the opposite direction (antiport, see Fig. 19.12 , [18] ). Movement of the pumped ion down its electrochemical gradient is by facilitated diffusion. The purpose of both types of co-transport is to use the energy in an electrochemical gradient to drive the movement of another solute against its gradient. An example of symport is the SGLT1 (sodium-glucose transport protein-1) in the intestinal epithelium [20] . SGLT1 uses the energy in a downhill transmembrane movement of Na + to transport glucose across the apical membrane against an uphill glucose gradient so that the sugar can be transported into the bloodstream.
4.4. Bacterial Lactose Transport
The secondary active symport system for lactose uptake in Escherichia coli is shown in Fig. 19.16 [21] . Lactose uptake is driven through a channel by a H + gradient generated by the bacterial electron transport system [22] . The free energy equation for transport described above ( Δ G = R T ln [ s o ′ ] / [ s o ] + Z F Δ Ψ ) can be rearranges for cases employing H + gradients (see Chapter 18) to:
Where Δ μ H + is the proton motive force; ΔΨ is the transmembrane electrical potential; R is the gas constant; T is the temperature in °K; n is the solute charge (+1 for protons); F is the Faraday; ΔpH is the transmembrane pH gradient.

Lactose transport system in Escherichia coli [21] . Uptake of lactose is coupled to the movement of an H + down its electrochemical gradient. This is an example of active transport, co-transport, and active.
It is the force on an H + (called the proton motive force) that drives lactose uptake. Note that the ability to take up lactose is a combination of the electrical gradient and the pH gradient. Although lactose uptake is directly coupled to H + transmembrane movement, it is possible to take up lactose even if the pH gradient is zero (ie, if the ΔΨ is sufficiently large).
4.5. Vectorial Metabolism, Group Translocation
Over 50 years ago, Peter Mitchell (see Chapter 18, Fig. 18.26) recognized the importance of what he termed “vectorial metabolism” [23] , [24] . Water-soluble enzymes convert substrate to product without any directionality. Mitchell proposed that many enzymes are integral membrane proteins that have a specific transmembrane orientation. When these enzymes convert substrate to product they do so in one direction only. This enzymatic conversion is therefore unidirectional, or “vectorial.” Mitchell expanded this basic concept into his now famous “chemiosmotic hypothesis” for ATP synthesis in oxidative phosphorylation (Chapter 18) [25] , [26] . For this revolutionary idea Mitchell was awarded the 1997 Nobel Prize in Chemistry.
Vectorial metabolism has been used to describe the mechanism for several membrane transport systems. For example, it has been reported in some cases the uptake of glucose into a cell may be faster if the external source of glucose is sucrose rather than free glucose. Through a vectorial transmembrane reaction, membrane-bound sucrase may convert external sucrose into internal glucose plus fructose more rapidly than the direct transport of free glucose through its transport system.
Mitchell defined one type of vectorial transport as group translocation, the best example being the PTS (phosphotransferase system) discovered by Saul Roseman in 1964.
PTS is a multicomponent active transport system that uses the energy of intracellular phosphoenol pyruvate (PEP) to take up extracellular sugars in bacteria. Transported sugars include glucose, mannose, fructose, and cellobiose. Components of the system include both plasma membrane and cytosolic enzymes. PEP is a high-energy phosphorylated compound (Δ G of hydrolysis is −61.9 kJ/mol) that drives the system. The high-energy phosphoryl group is transferred through an enzyme bucket brigade from PEP to glucose producing glucose-6-phosphate in several steps (PEP → EI → HPr →EIIA → EIIB → EIIC → glucose-6-phosphate). The sequence is depicted in more detail in Fig. 19.17 [27] . HPr stands for heat-stable protein that carries the high-energy ∼P from EI (enzyme-I) to EIIA. EIIA is specific for glucose and transfers ∼P to EIIB that sits next to the membrane where it takes glucose from the transmembrane EIIC and phosphorylates it producing glucose-6-phosphate. Although it is glucose that is being transported across the membrane, it never actually appears inside the cell as free glucose but rather as glucose-6-phosphate. Free glucose could leak back out of the cell via a glucose transporter, but glucose-6-phosphate is trapped inside the cell where it can rapidly be metabolized through glycolysis. Group translocation is defined by a transported solute appearing in a different form immediately after crossing the membrane.

The bacterial PTS system for glucose transport [27] .
5. Ionophores
The term ionophore means “ion bearer.” Ionophores are small, lipid-soluble molecules, usually of microbial origin, whose function is to conduct ions across membranes [28] , [29] . They are facilitated diffusion carriers that transport ions down their electrochemical gradient. Ionophores can be divided into two basic classes: channel formers and mobile carriers ( Fig. 19.18 ) [30] . Channel formers are long lasting, stationary structures that allow many ions at a time to rapidly flow across a membrane. Mobile carriers bind to an ion on one side of a membrane, dissolve in and cross the membrane bilayer and release the ion on the other side. They can only carry one ion at a time. Four representative ionophores will be discussed: the K + ionophore valinomycin, the proton ionophore 2,4-dinitrophenol, synthetic crown ethers, and the channel-forming ionophore nystatin ( Fig. 19.19 ).

Two basic types of ionophores: channel formers (left) and mobile carriers (right) [30] .

Representative examples of ionophores: the K + ionophore valinomycin, the proton ionophore 2,4-dinitrophenol, the synthetic crown ether 18-crown-6, and the channel forming ionophore nystatin.
5.1. Valinomycin
Superficially valinomycin resembles a cyclic peptide ( Fig. 19.19 ). However, upon closer examination the ionophore is actually a 12-unit (dodeca) depsi peptide where amino acid peptide bonds are alternated with amino alcohol ester bonds. Therefore the linkages that hold the molecule together alternate between nitrogen esters (peptide bonds) and oxygen esters. The units that comprise valinomycin are d - and l -valine (hence the name “valinomycin”), hydroxyvaleric acid and l -lactic acid. The circular structure is a macrocyclic molecule with the 12 carbonyl oxygens facing the inside of the ring where they chelate a single K + . The outside surface of valinomycin is coated with nine hydrophobic side chains of d - and l -valine and l -hydroxyvaleric acid. The polar interior of valinomycin precisely fits one K + . The binding constant for K + -valinomycin is 10 6 while Na + -valinomycin is only 10. This emphasizes the high selectivity valinomycin has for K + over Na + . Valinomycin, therefore, has an oily surface that readily dissolves in a membrane lipid bilayer, carrying K + across the membrane down its electrochemical gradient.
Valinomycin was first recognized as a potassium ionophore by Bernard Pressman in the early 1960's [31] , [32] . He reported that valinomycin, a known antibiotic, stimulated K + uptake and H + efflux from mitochondria. Many studies showed that valinomycin dissipates essential transmembrane electrochemical gradients causing tremendous metabolic upheaval in many organisms including microorganisms. It is for this reason that valinomycin was recognized as an antibiotic long before it was identified as an ionophore. Currently several ionophores are added to animal feed as antibiotics and growth enhancing additives [33] . Recently valinomycin has been reported to be the most potent agent against SARS-CoV (severe acute respiratory-syndrome coronavirus), a severe form of pneumonia first identified in 2003 [34] .
5.2. 2,4-Dinitrophenol
2,4-Dinitrophenol (DNP, Fig. 19.19 ) is considered to be the classic uncoupler of oxidative phosphorylation (see Chapter 18). It is a synthetic lipid-soluble proton ionophore that dissipates proton gradients across bioenergetic membranes (mitochondrial inner, thylakoid, bacterial plasma). An uncoupler is therefore an H + -facilitated diffusion carrier. Elucidating the role of DNP in uncoupling oxidative phosphorylation was an essential component in support of Peter Mitchell's chemiosmotic hypothesis [25] . Electron movement from NADH or FADH 2 to O 2 via the mitochondrial electron transport system generates a considerable amount of electrical energy that is partially captured as a transmembrane pH gradient (see Chapter 18). The movement of H + s back across the membrane, driven by the electrochemical gradient, is through a channel in the F 1 ATPase (an F-type primary active transport system discussed above, (Chapter 19, Section 4.1 )) that is coupled to ATP synthesis. DNP short-circuits the H + gradient before it can pass through the F 1 ATPase, thus uncoupling electron transport, the energy source for the H + gradient, from ATP synthesis. Therefore, in the presence of DNP, electron transport continues, even at an accelerated rate, but ATP production is diminished. The energy that should have been converted to chemical energy in the form of ATP is then released as excess heat.
This combination of properties led to the medical application of DNP to treat obesity from 1933 to 1938 [35] . Upon addition of DNP:
- • The patient became weak due to low ATP levels.
- • Breathing increased due to increased electron transport to rescue ATP production.
- • Metabolic rate increased.
- • Body temperature increased due to inability to trap electrical energy as chemical energy in the form of ATP, releasing heat.
- • Body weight decreased due to increased respiration burning more stored fat.
DNP was indeed a successful weight loss drug. Two of the early proponents of DNP use as a diet drug, Cutting and Tainter at Stanford University, estimated that more than 100,000 people in the United States had tested the drug during its first year in use [35] . DNP, however, did have one disturbing side effect—death! Fatality was not caused by a lack of ATP, but rather by a dangerous increase in body temperature (hyperthermia). In humans, 20–50 mg/kg of DNP can be lethal. Although general use of DNP in the United States was discontinued in 1938, it is still employed in other countries and by bodybuilders to eliminate fat before competitions.
5.3. Crown Ethers
Crown ethers are a family of synthetic ionophores that are generally similar in function to the natural product valinomycin [36] . The first crown ether was synthesized by Charles Pederson ( Fig. 19.20 ) while working at DuPont in 1967. For this work Pedersen was co-awarded the 1987 Nobel Prize in Chemistry. Crown ethers are cyclic compounds composed of several ether groups. The most common crown ethers are oligomers of ethylene oxide with repeating units of (—CH 2 CH 2 O—) n where n = 4 (tetramer), n = 5 (pentamer), or n = 6 (hexamer). Crown ethers are given structural names, X -crown- Y , where X is the total number of atoms in the ring and Y is the number of these atoms that are oxygen. Crown refers to the crown-like shape the molecule takes. Crown ether oxygens form complexes with specific cations that depend on the number of atoms in the ring. For example, 18-crown-6 ( Fig. 19.19 ) has high affinity for K + , 15-crown-5 for Na + , and 12-crown-4 for Li + . Like valinomycin, the exterior of the ring is hydrophobic, allowing crown ethers to dissolve in the membrane lipid bilayer while carrying the sequestered cation down its electrochemical gradient. It is now possible to tailor make crown ethers of different sizes that can encase a variety of catalysts for phase transfer into the bilayer hydrophobic interior where they can be used to catalyze reactions inside the membrane.

Charles Pedersen, 1904–1989.
5.4. Nystatin
Nystatin ( Fig. 19.19 ) is a channel-forming ionophore that creates a hydrophobic pore across a membrane [37] , [38] . Channel-forming ionophores allow for the rapid facilitated diffusion of various ions that depend on the dimensions of the pore. Nystatin, like other channel-forming ionophores (eg, amphotericin B and natamycin), is a commonly used antifungal agent. Finding medications that can selectively attack fungi in the presence of normal animal cells presents a difficult challenge since both cell types are eukaryotic. Bacteria, being prokaryotes, are sufficiently different to present a variety of anti-bacterial approaches not amenable to fungi. However, fungi do have an Achilles heel. Fungal plasma membranes have as their dominant sterol ergosterol, not the animal sterol cholesterol (see Chapter 5). Nystatin binds preferentially to ergosterol, thus targeting fungi in the presence of animal cells. When present at sufficient levels, nystatin complexes with ergosterol and forms transmembrane channels that lead to K + leakage and death of the fungus. Nystatin is a polyene antifungal ionophore that is effective against many molds and yeast including Candida . A major use of nystatin is as a prophylaxis for AIDS patients who are at risk for fungal infections.
6. Gap Junctions
Gap junctions are a common structural feature of many animal plasma membranes [39] , [40] . In plants similar structures are known as plasmodesmata. Gap junctions were introduced earlier in Chapter 11 (see Fig. 11.6). Gap junctions represent a primitive type of intercellular communication that allows transmembrane passage of small solutes like ions, sugars, amino acids, and nucleotides while preventing migration of organelles and large polymers like proteins and nucleic acids. Gap junctions connect the cytoplasms of two adjacent cells through nonselective channels. Connections through adjacent cells are at locations where the gap between cells is only 2–3 nm. This small gap is where the term “gap junction” originated. Gap junctions are normally clustered from a few to over a 1000 in select regions of a cell plasma membrane.
Early experiments involved injecting fluorescent dyes, initially fluorescein (molecular weight 300), into a cell and observing the dye movement into adjacent cells with a fluorescence microscope [41] , [42] . Currently Lucifer Yellow has become the fluorescent dye of choice for gap junction studies, replacing fluorescein. At first, the dye only appeared in the initially labeled cell. With time, however, the dye was observed to spread to adjacent cells through what appeared to be points on the plasma membrane. These points were later recognized as gap junctions. By varying the size of the fluorescent dye, it was shown that there was an upper size limit for dye diffusion. Solutes had to have a molecular weight of less than ∼1200 to cross from one cell to another [41] .
Although gap junctions were obviously channels that connected the cytoplasms of adjacent cells, it was years before their structure, shown in Fig. 19.21 , was determined [43] , [44] . Each channel in a gap junction is made up of 12 proteins called connexins. Six hexagonally arranged connexins are associated with each of the adjacent cell plasma membranes that the gap junction spans. Each set of six connexins is called a connexon and forms half of the gap junction channel. Therefore, one gap junction channel is composed of 2 aligned connexons and 12 connexins. Each connexin has a diameter of about 7 nm and the hollow center formed between the 6 connexins (the channel) is about 3 nm in diameter. Gap junctions allow adjacent cells to be in constant electrical and chemical communication with one another. Of particular importance is the rapid transmission of small second messengers, such as inositol triphosphate (IP 3 ) and Ca 2+ .

Gap junction [43] . Six connexins form a connexon and one connexon from each cell unite to form a gap junction.
It appears that all cells in the liver are interconnected through gap junctions. This presents a possible dilemma. If even a single cell is damaged, deleterious effects may be rapidly spread throughout the entire liver. Preventing this is one important function of Ca 2+ . Extracellular Ca 2+ is ∼10 −3 mol/L while intracellular levels are maintained at ∼10 −6 mol/L. If a cell is damaged, Ca 2+ rushes in, dramatically increasing intracellular Ca 2+ . Gap junction channels close if intracellular Ca 2+ reaches 10 −3 mol/L, thus preventing the spread of damage.
Gap junctions are particularly important in cardiac muscle as the electrical signals for contraction are passed efficiently through these channels [45] . As would be expected, malfunctions of gap junctions lead to a number of human disorders including demyelinating neurodegenerative diseases, skin disorders, cataracts, and even some types of deafness.
7. Other Ways to Cross the Membrane
There are several other ways that solutes, including large macromolecules, can cross membranes. These methods include receptor-mediated endocytosis (RME, discussed in Chapter 17), phagocytosis, pinocytosis, exocytosis, and membrane blebbing. These methods involve large sections of a membrane containing many lipids and proteins.
Two similar transport processes that have been known for a long time are pinocytosis and phagocytosis [46] . Both involve nonspecific uptake (endocytosis) of many things from water and ions through to large macromolecules and, for phagocytosis, even whole cells. Pinocytosis is Greek for “cell drinking” and involves the plasma membrane invaginating a volume of extracellular fluid and anything it contains including water, salts, biochemicals and even soluble macromolecules. Phagocytosis is Greek for “cell eating” and involves the plasma membrane invaginating large insoluble solids.
7.1. Pinocytosis
Pinocytosis is a form of endocytosis involving fluids containing many solutes. In humans, this process occurs in cells lining the small intestine and is used primarily for absorption of fat droplets. In endocytosis the cell plasma membrane extends and folds around desired extracellular material, forming a pouch that pinches off creating an internalized vesicle ( Fig. 19.22 , [19] , [20] , [21] , [22] , [23] , [24] , [25] , [26] , [27] , [28] , [29] , [30] , [31] , [32] , [33] , [34] , [35] , [36] , [37] , [38] , [39] , [40] , [41] , [42] , [43] , [44] , [45] , [46] , [47] ). The invaginated pinocytosis vesicles are much smaller than those generated by phagocytosis. The vesicles eventually fuse with the lysosome whereupon the vesicle contents are digested. Pinocytosis involves a considerable investment of cellular energy in the form of ATP and so is many 1000 times less efficient than RME (see Chapter 17). Also, in sharp contrast to RME, pinocytosis is nonspecific for the substances it accumulates. Pinocytosis is not a recent discovery as it was first observed decades before the other transport systems discussed in Chapter 19. Its discovery is attributed to Warren Lewis in 1929.

Pinocytosis, a type of endocytosis. An invagination of the plasma membrane encapsulates many water-soluble solutes ranging in size from salts to macromolecules.
7.2. Phagocytosis
Phagocytosis is a type of endocytosis that involves uptake of large solid particles, often >0.5 mm [47] . The particles are aggregates of macromolecules, parts of other cells, and even whole microorganisms and, in contrast to pinocytosis (shown in Fig. 19.22 ), phagocytosis has surface proteins that specifically recognize and bind to the solid particles. Fig. 19.23 [48] depicts events in phagacytosis. Phagocytosis is a routine process that ameba and ciliated protozoa use to obtain food. In humans, phagocytosis is restricted to specialized cells called phagocytes that include white blood cell neutrophils and macrophages. As with pinocytosis, phagocytosis generates intracellular vesicles called phagosomes that have sequestered solid particles they transport to the lysosome for digestion. Phagocytosis is a major mechanism used by the immune system to remove pathogens and cell debris. In fact, very early studies of the immune system led Elie Metchnikoff to discover phagocytosis in 1882. For this work Metchnikoff shared the 1908 Nobel Prize in Medicine with Paul Ehrlich.

Phagocytosis, a type of endocytosis that involves uptake of large solid particles.
7.3. Exocytosis
Exocytosis is the process by which cells excrete waste and other large molecules from the cytoplasm to the cell exterior [49] and therefore is the opposite of endocytosis. Exocytosis generates vesicles referred to as secretory or transport vesicles (Chapter 17). In exocytosis, intracellular (secretory) vesicles fuse with the plasma membrane and release their aqueous sequestered contents to the outside at the same time that the vesicular membrane hydrophobic components (mostly lipids and proteins) are added to the plasma membrane ( Fig. 19.24 , [50] ). Steady state composition of the plasma membrane results from a balance between endocytosis and exocytosis. The resultant process of plasma membrane recycling is amazingly fast. For example, pancreatic secretory cells recycles an amount of membrane equal to the whole surface of the cell in ∼90 min. Even faster are macrophages that can recycle contents of their plasma membrane in only 30 min.

Exocytosis. Intracellular secretory vesicles fuse with the plasma membrane releasing their water-soluble contents to the outside and adding membrane material to the plasma membrane [50] .
Before approaching the plasma membrane for fusion, exocytosis vesicles had a prior life that is considered in Chapter 17. The vesicles must first dock with the plasma membrane, a process that keeps the two membranes separated by <5–10 nm. During docking, complex molecular rearrangements occur to prepare the membranes for fusion. The process of vesicle fusion and release of aqueous compartment components is driven by SNARE proteins (see Chapters 10 and 17Chapter 10Chapter 17) [51] , [52] .
7.4. Blebbing
Blebbing of the plasma membrane is a morphological feature of cells undergoing late stage apoptosis (programmed cell death, see Chapter 24) [53] . A bleb is an irregular bulge in the plasma membrane of a cell caused by localized decoupling of the cytoskeleton from the plasma membrane. The bulge eventually blebs off from the parent plasma membrane taking part of the cytoplasm with it. It is clear in Fig. 19.25 [54] that the plasma membrane of an apoptotic cell is highly disintegrated and has lost the integrity required to maintain essential transmembrane gradients. Blebbing is also involved in some normal cell processes, including cell locomotion and cell division.

Membrane blebbing during apoptosis [54] .
Carefully controlled solute movement into and out of cells is an essential feature of life. There are many ways solutes are transported across the thin (∼40 Å) membrane hydrophobic barrier. Transport is divided into passive diffusion and active transport. A biological membrane is semipermeable, being permeable to some molecules, most notably water (osmosis), while being very impermeable to most solutes that require some form of transporter. Passive diffusion (simple and facilitated) only requires the energy inherent in the solute's electrochemical gradient and results in its equilibrium across the membrane. In contrast, active transport requires additional energy (ie, ATP), and results in a nonequilibrium, net accumulation of the solute. Passive transport can involve simple diffusion or facilitated carriers including ionophores and channels. Active transport comes in many, often complex forms. Examples of active transport include primary active transport (uniport), secondary active transport (co-transport, antiport), and group translocation. Besides the multitude of transport systems, transport can be accomplished by gap junctions, receptor mediated endocytosis, phagocytosis, pinocytosis, exocytosis, and apoptotic membrane blebbing.
Chapter 20 will discuss bioactive lipids, highly specialized lipids that are functional at very low levels. Discussed bioactive lipids include ceramides, diacylglycerol, eicosanoids, steroid hormones, and phosphatidic acid.
An Open and Shut Case: Membrane Transport in Health and Disease
4 Feb 2019 | Robert Wilkins

There are three types of protein in cells that can be the target for drugs: receptors, enzymes and transporters. While inhibitors of receptor proteins (‘Beta blockers’) and inhibitors of phosphodiesterase enzymes (‘Viagra’) are well known, the function of membrane transport proteins is less familiar but equally important.
My interest in membrane transport proteins was sparked when I first learnt about them as an undergraduate over a quarter of a century ago. From almost the first lecture, transport proteins featured regularly and it rapidly became clear that they had a fundamental role in physiological function. One of my tutors was an expert in the field of membrane transport, so our discussions in tutorials regularly led us to consider what these proteins do and how they do it. Enthused by those discussions, my work at Finals focused on transporters and I had my first taste of laboratory research, characterising the systems regulating the levels of acid in cartilage cells. A D.Phil building on that work followed, leading to a research career that has investigated the possible involvement of membrane transport proteins in two disease states: arthritis and cancer. We’ll come back to that work at the end of this blog, but first let me explain the basics of membrane transport…
The 35 trillion cells in the human body vary greatly in appearance, location and function, but they all possess a membrane, which separates the fluids inside and out. The membrane is a flexible ‘sea’ of lipid molecules, in which float a variety of protein molecules.
Some of the proteins act as markers (‘recognition signals’) to identify the cell type; others are docking stations (‘receptors’) for chemicals such as hormones and neurotransmitters that instruct the cell what to do; some are enzymes, catalysing reactions that create chemical messages to convey those instructions; yet others act as transporters ( ‘channels’ and ‘carriers’ ) to allow for movement of solutes (ions and organic molecules such as glucose) between the cell and its surroundings.
This last category is needed because most of the chemicals relevant to cell function don’t readily cross the lipid membrane. While gases such as oxygen and carbon dioxide can dissolve in the lipid membrane and so move into or out of the cell, metabolites and ions need assisted passage. Membrane transport proteins therefore play essential roles in cellular function: their actions underpin nerve impulses, muscle contraction, the heartbeat, urine formation and absorption of food in the gut. Perhaps unsurprisingly, inadequate or excess activity of these proteins leads to disease, while a number of common drugs exert their actions by modifying transporter function.

Channels are like doors…
Transporters are effectively ‘doors’ in the ‘wall’ that is the cell membrane, through which solutes can move. The simplest transporters – leak channels – are like a door frame that lacks a door connecting the two sides of the membrane. Solutes move from the side where they are plentiful to the side where they are in short supply, but no control can be exerted over how much traffic occurs across the membrane. Malaria parasites in infected red blood cells may subvert these channels to establish a pathway to gain nutrients from outside the cell and expel the waste products of their metabolism.
In a gated channel, there is a door within the frame that can be open or closed, providing regulated traffic. The opening of ion channels sensitive to voltage is fundamental to nervous signaling and the electrical activity that creates the heartbeat: defects in the function of voltage-gated channels in the heart can lead to cardiac arrhythmias. Channels that are gated by neurotransmitter binding allow nerve-to-nerve signaling and convert nervous impulses into muscle contractions, while the opening of channels when the membrane of tendon cells is stretched initiates the knee jerk reflex.
Other channels control the secretion of saliva and of mucus in the lung. In the genetic disease cystic fibrosis, a channel protein that allows the passage of chloride ions is faulty. The movement of chloride is needed to draw water into the airway, without which the protective lining is too sticky and the airway becomes prone to infection.

…and carriers are like revolving doors
Carrier proteins function like revolving doors and, just as we can be conveyed through a revolving door by someone pushing from behind or coming in the opposite direction, so some of these transporters can use the movement of one solute to drive the flow of another against its natural tendency, so leading to accumulation inside, or extrusion from, the cell. Many of these transporters fulfil ‘ housekeeping ‘ functions for cells, exporting wastes and maintaining cell size. Red blood cells in sickle cell disease patients show abnormal housekeeping, which increases the likelihood that the cells will clump in blood capillaries. Transporters of this type are essential for scavenging glucose and amino acids in the gastrointestinal tract: defects lead to nutrient malabsorption. In the kidney, their action allows the body to vary the composition of the urine in response to altered salt and water intake. ‘Water tablets’ (aka diuretics such as furosemide and chlorthiazide) exert their effects by inhibiting carriers that move sodium ions across the membranes of renal cells. Some antidepressants (for example, ‘Prozac’) work by inhibiting transporters for neurotransmitters, increasing levels of these signalling molecules in the brain to alter mood; cocaine also exerts some of its actions through effects on transmitter transporters.
The most complicated transporters are like electrically operated revolving doors, where the door rotates without us needing to push on it. These ‘active pumps’ use the cell’s energy store to power ion movements and create asymmetric distributions of ions on either side of the cell membrane. The asymmetry creates gradients and, when ions move down those gradients on carriers, the ‘push’ on the door provides the drive that enables other solutes to be accumulated or expelled as already described. This powered transport also underlies the secretion of acid in the stomach, which is important for the digestion of ingested proteins: the drug omeprazole, used to treat gastric reflux and ulcers, inhibits this pump.
Arthritis and Cancer
My research has focused on two proteins that perform housekeeping roles in cells and their possible involvement in disease. The first protein is the Na+-H+ exchanger (NHE), which is one of the transporters that regulates the levels of acid (the pH) in cells. In cartilage, chondrocytes are responsible for the upkeep of the extracellular matrix that surrounds them and acts as a cushion between the bones in joints. My work showed that the level of acid inside chondrocytes influences the production of new cartilage. I also showed that chondrocytes rely almost exclusively on NHE to regulate their pH. Therefore, anything that changes the activity of this protein can lead to altered levels of cartilage synthesis through changes in pH: chemicals that are found at higher levels in arthritic joints have such effects. This marks out the carrier protein as a potential pharmaceutical target to promote synthesis of new cartilage.
The second protein is the K+-Cl- cotransporter (KCC), which is one of the transporters by which cells regulate their volume. With colleagues in Oxford and Taiwan, I have investigated how cancer cells use KCC to defend themselves from swelling and bursting when water enters them. Increased numbers of KCC proteins in cancer cells promotes the proliferation and invasion of these cells in tissues such as the cervix. Understanding how KCC does that could open up new approaches for limiting cancer development and spread in those tissues.
These are just two examples of the ways in which this array of proteins, with diverse distribution and function in the body, are being recognised as targets for drugs, prospects for gene therapy, and even as possible routes of drug delivery in the intestine.
Read more on our blog

Voices from the past: The surprising stories told by medieval German nuns
20 Jun 2024

“I have receiv’d by the hands of mr Fleming”: Thomas Smith, the Fleming Family and St Edmund Hall
22 May 2024
Category: Research
Blog Categories
- Library, Arts & Archives
View All Blog Posts

Robert Wilkins
Professor Robert Wilkins holds the American Fellowship in Physiology at St Edmund Hall. He oversees the admission, teaching and pastoral support of Biomedical Sciences and Medicine students and provides tutorials covering cellular and systems physiology.
View profile
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Open access
- Published: 11 April 2024
A CRISPRi/a screening platform to study cellular nutrient transport in diverse microenvironments
- Christopher Chidley ORCID: orcid.org/0000-0002-8212-3148 1 ,
- Alicia M. Darnell 2 ,
- Benjamin L. Gaudio 1 ,
- Evan C. Lien 2 ,
- Anna M. Barbeau 2 , 3 ,
- Matthew G. Vander Heiden ORCID: orcid.org/0000-0002-6702-4192 2 , 3 , 4 &
- Peter K. Sorger ORCID: orcid.org/0000-0002-3364-1838 1 , 5
Nature Cell Biology volume 26 , pages 825–838 ( 2024 ) Cite this article
9600 Accesses
4 Citations
37 Altmetric
Metrics details
- Cancer metabolism
- Functional genomics
- Systems biology
Blocking the import of nutrients essential for cancer cell proliferation represents a therapeutic opportunity, but it is unclear which transporters to target. Here we report a CRISPR interference/activation screening platform to systematically interrogate the contribution of nutrient transporters to support cancer cell proliferation in environments ranging from standard culture media to tumours. We applied this platform to identify the transporters of amino acids in leukaemia cells and found that amino acid transport involves high bidirectional flux dependent on the microenvironment composition. While investigating the role of transporters in cystine starved cells, we uncovered a role for serotonin uptake in preventing ferroptosis. Finally, we identified transporters essential for cell proliferation in subcutaneous tumours and found that levels of glucose and amino acids can restrain proliferation in that environment. This study establishes a framework for systematically identifying critical cellular nutrient transporters, characterizing their function and exploring how the tumour microenvironment impacts cancer metabolism.
Similar content being viewed by others

Nutrient transporters: connecting cancer metabolism to therapeutic opportunities

New aspects of amino acid metabolism in cancer

Cancer metabolism: looking forward
Altered cellular metabolism is a common feature of tumours, enabling cancer cells to maximize proliferation in nutrient-limited environments 1 . Cellular adaptations to the tumour microenvironment (TME) generally include an increase in nutrient uptake, a diversification of uptake mechanisms and a rewiring of metabolism so tumours can more efficiently convert available nutrients into biomass 2 . Amino acids contribute substantially to the formation of cellular biomass 3 , and many cancers are dependent on environmental supplies of non-essential amino acids for growth and survival. For example, acute lymphoblastic leukaemias are dependent on asparagine, luminal breast cancers and breast cancer brain metastases exhibit serine auxotrophy, and certain cancers silence arginine biosynthesis or upregulate glutamine metabolism 2 , 4 , 5 , 6 , 7 . Such dependencies can potentially be targeted therapeutically by inhibiting nutrient uptake 8 .
Nutrients are actively transported across cell membranes by a large class of transporter proteins called solute carriers (SLCs) 9 . The activity of many SLCs is influenced by the composition of the microenvironment 10 . Studying the function of these SLCs, and identifying potential therapeutic targets, requires characterizing transport under physiologically relevant conditions. Much of our current knowledge derives from experiments in cell-free systems, and many SLC substrates identified in these experiments (hereafter ‘annotated’ substrates) have not been evaluated in cells or organisms 11 . For example, over 60 SLCs are annotated as amino acid transporters, but it is unclear which are dominant or growth-limiting in cells 11 , 12 , 13 . Recent studies have uncovered the transporters for essential metabolites such as NAD + (refs. 14 , 15 , 16 ), choline 17 and glutathione 18 , 19 , but 20–30% of SLCs, including many that are essential in human cells, have no identified function 9 , 20 . The development of clustered regularly interspaced short palindromic repeats (CRISPR) screening 21 and efforts to better characterize the composition of the TME 22 , 23 present opportunities to better define the role of nutrient transporters in human cells.
In this Resource, we describe the use of CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) screening using custom single guide RNA (sgRNA) libraries to systematically interrogate the contributions to nutrient uptake or release of all SLC and ATP-binding cassette (ABC) transporters found in human cells. We screen for transporters responsible for amino acid import in K562 chronic myelogenous leukaemia cells and assess transporter function using mass spectrometry-based transport assays. We also use CRISPRi to identify transporters essential for cell proliferation in standard and physiological growth media, and in subcutaneous tumours in mice. Finally, we use CRISPRa to identify nutrients limiting for proliferation across environments, providing a framework for characterizing cancer-relevant nutrient transporters and how their interaction with the TME shapes metabolism.
Cell-based screens for amino acid transporters
To assess transporter function, we employed a dual approach: transporter knockdown (KD) via CRISPRi and overexpression (OE) via CRISPRa. Given previous findings that transporter genes are often top hits in CRISPR screens 21 , 24 , 25 , 26 , we hypothesized that KD/OE would alter substrate transport rates and that such changes could be isolated in growth-based pooled screens (Fig. 1a ). Specifically, in conditions where a nutrient limits proliferation, KD of a transporter needed for its import would reduce proliferation and, conversely, OE of a transporter capable of importing that nutrient would increase proliferation. Transporters involved in nutrient export would exhibit opposite phenotypes compared with importers.

a , A cartoon of the general approach used to identify transporters in cells. Individual transporter genes are knocked down via CRISPRi or overexpressed via CRISPRa, and modified transport activity is detected by changes in proliferation. b , CRISPRi/a of transporters leads to specific changes in gene expression. Expression levels in K562 CRISPRi/a cells with specific or non-targeting control (NTC) sgRNAs were quantified by RT–qPCR relative to the housekeeping gene GAPDH. Data are mean ± s.e.m of n = 3 technical replicates. The horizontal dashed lines represent the average of both NTCs. n.d., not detected. c , CRISPRi/a of transporters leads to changes in protein level at the plasma membrane. K562 CRISPRi/a cells were incubated with a cell-impermeable biotinylation reagent, and plasma membrane proteins were isolated by streptavidin affinity purification and analysed by western blotting. d , The identification of amino acids that limit proliferation of K562 cells when their level in growth medium is reduced. Data were determined using a luminescent cell viability assay and represent the average log 2 FC relative to time of 0 days (T0) of n = 4 biologically independent samples. e , A cartoon of the pooled screening strategy used to identify amino acid transporters. Library pools were grown in RPMI media where specific amino acids were present at a level that reduced proliferation by 50% relative to complete RPMI (low amino acid). a.a., amino acid. f , Volcano plots of transporter CRISPRi/a screens in K562 cells in low lysine and low arginine. Black circles represent transporter genes, and red circles represent negative control genes. n = 2 screen replicates. g , Bubble plots displaying CRISPRi screen scores determined for all 64 transporters annotated as capable of amino acid transport 12 . h , Same as g for CRISPRa screens. In f – h , the phenotype scores represent averaged and normalized sgRNA enrichments in low amino acid versus RPMI, and −log 10 ( P value) was determined using a Mann–Whitney test of sgRNA enrichments compared with all NTC sgRNAs. Source numerical data and unprocessed blots are available in Source data .
Source data
To validate this system, we targeted four expressed SLC7 amino acid transporters (SLC7A1, SLC7A5, SLC7A6 and SLC7A8) and three not expressed (SLC7A2, SLC7A3 and SLC7A7) in K562 cells expressing either dCas9-KRAB (CRISPRi) or dCas9-SunTag (CRISPRa). CRISPRi/a of transporters resulted in strong and specific changes in messenger RNA expression (Fig. 1b ). SLC7A5, SLC7A6, SLC7A7 and SLC7A8 transporters function as heterodimers with SLC3A2. Quantitative reverse transcription polymerase chain reaction (RT–qPCR) analysis revealed upregulation of SLC3A2 levels in SLC7A5, SLC7A7 and SLC7A8 CRISPRa cells, consistent with higher levels of functional heterodimers (Fig. 1b ). Changes in cell surface protein levels in these cells mirrored RNA-level changes (Fig. 1c ). We conclude that CRISPR-based perturbation of SLCs resulted in the anticipated changes in protein levels.
Next, we constructed CRISPRi and CRISPRa sgRNA pooled lentiviral libraries targeting 489 annotated members of the SLC and ABC transporter families (hereafter ‘transporters’) 20 , 27 , 28 ; the library includes 10 sgRNAs/gene and 730 non-targeting control (NTC) sgRNAs (Extended Data Fig. 1a and Supplementary Table 1 ). The primary function of ABC transporters is to export xenobiotics from cells, but some transporters export metabolites 29 , 30 , suggesting a role in nutrient homeostasis. Libraries of single transporter KD or OE cells were prepared by transduction of K562 CRISPRi or CRISPRa parental lines (Extended Data Fig. 1b ).
To enable selection for transporter phenotypes, we identified all amino acids in RPMI-1640 culture medium (RPMI) whose absence would limit proliferation (Fig. 1d ). For 13 of the 19 amino acids in RPMI, reduced levels decreased proliferation, indicating net consumption of these amino acids by cells 3 . Asn, Asp, Glu, Gly and Pro removal from the medium did not impact proliferation, precluding transporter identification for these amino acids. K562 cells initially responded to low Ser but proliferation returned to normal, probably due to upregulation of serine biosynthesis enzymes 31 . We were therefore only able to screen for serine transporters under transient and mild growth limitation. Cystine (the oxidized conjugate of Cys most abundant in culture medium) deprivation induced substantial cell death, through ferroptosis 32 , 33 .
We performed pooled CRISPRi/a screens at amino acid concentrations that reduced K562 proliferation by ~50% for all 13 growth-limiting amino acids to identify the dominant and growth-limiting amino acid transporters (Supplementary Tables 2 and 3 ). Libraries of single transporter KD or OE cells underwent three rounds of low amino acid exposure followed by one recovery day in complete medium (Fig. 1e ). Changes in library content were compared with a control screen in complete RPMI medium, and scores were calculated for each transporter 25 , 34 ( Methods ).
Amino acid limitation activates the GCN2 starvation response and represses mTOR activity, resulting in changes in transporter gene expression 12 . To assess the extent to which this occurred under screening conditions we used western blotting to assay phosphorylation of mTORC1 and GCN2 targets (Extended Data Fig. 1c ). K562 cells cultured in low Arg, Lys or His screening conditions showed mild GCN2 activation and mTOR repression. Refreshing the medium after 1 day incubation relieved those effects in low amino acid conditions, but not in complete starvation conditions (Extended Data Fig. 1c ). We investigated if low amino acid conditions altered transporter gene expression; low-Arg and low-Leu screening conditions increased expression of most genes in our SLC7 transporter panel, but not all of them, consistent with results for HEK293T cells in single amino acid dropout media 35 . Critically, however, despite changes in transporter expression caused by screening media, CRISPRi/a of SLC7 genes resulted in additional and specific strong up- or downregulation of mRNA levels and concomitant changes in protein levels at the plasma membrane (Extended Data Figs. 1d,e and 2a ). We conclude that while low amino acid screening conditions induce a slight upregulation in the expression of transporters, this does not interfere with the identification of specific phenotypes resulting from transporter CRISPRi/a.
CRISPRi/a screening results
We found that CRISPRi screens in low amino acid conditions mainly found hits with negative phenotype scores, consistent with a role for these transporters in net amino acid import. For all 13 conditions, we identified at least one transporter with significantly depleted sgRNAs (Fig. 1f,g and Extended Data Fig. 2b ). Screens with one strong negative hit suggest that a single transporter is responsible for the bulk of the import of the limiting amino acid. For example, we found that SLC7A5 (LAT1) 36 is probably the primary importer for all large neutral amino acids in growth-limited conditions 12 , 28 . Screens with more than one negative hit suggest a role for multiple partially redundant transporters; this was observed for CRISPRi of SLC1A5, SLC38A1 and SLC38A2 in glutamine-limiting conditions (Fig. 1g ).
In contrast, CRISPRa screens in low amino acid conditions primarily yielded hits with positive scores, consistent with increased amino acid import (Fig. 1f,h and Extended Data Fig. 2b ). CRISPRa identified more hits than CRISPRi since it queries the role of all transporters, not only the ~50% (ref. 37 ) that are well expressed in K562 cells. For example, SLC7A1, SLC7A2 and SLC7A3 (CAT1, CAT2 and CAT3), annotated as arginine and lysine transporters, were strong hits in low-Lys and low-Arg CRISPRa screens. However, only SLC7A1 was a hit in those CRISPRi screens, as only SLC7A1 is expressed (Fig. 1b,f ). Thus, while SLC7A1 is the primary transporter for Lys and Arg in K562 cells, SLC7A2 and SLC7A3 are also capable of transport. CRISPRi/a screens were reproducible across biological replicates (Extended Data Fig. 2c,d ), and strong CRISPRi hits consistently appeared in corresponding CRISPRa screens, indicating that transporter activity is not saturated under screening conditions.
Our parallel screening across many low amino acids conditions enabled us to derive biological insight from negative results for transporters with significant phenotypes in at least one condition, demonstrating that CRISPRi/a was effective. Negative findings can be interpreted as evidence of the transporter’s lack of activity for those tested amino acids (Extended Data Fig. 2b ).
To delve deeper into transporter phenotypes, we constructed cell lines expressing a single sgRNA identified in screens. Transporter KD/OE was confirmed by RT–qPCR, and growth phenotypes were measured in competition assays (Extended Data Fig. 3a,b and Methods ). We validated screens results using seven SLC7 family genes having varying phenotypes in low His, Lys and Arg conditions. The validation closely aligned with screen outcomes, reinforcing the reliability of the approach (Fig. 2a ). For example, SLC7A7 OE increased proliferation in low Lys but not in low Arg in both screens and competition assays (Fig. 2a ). To explore the potential impact of substrate competition, we reduced Lys levels in RPMI and again observed no phenotype for SLC7A7 OE in low Arg. These data suggest that, at growth-limiting concentrations, SLC7A7 imports Lys but not Arg or any other tested amino acid (Fig. 2a ).

a , The phenotype scores obtained in transporter CRISPRi/a screens for SLC7 family genes were validated in competition assays in K562 cells in RPMI and in RPMI with amino acids adjusted to human plasma levels (PAA-RPMI). b , The import of amino acids into K562 cells in RPMI was determined by quantifying the intracellular accumulation of stable heavy-isotope-labelled amino acids over time by GC–MS. One example representative of six independent experiments. c , Amino acid import and cellular consumption rates of K562 cells growing in RPMI. Import rates were determined by linear regression of the early phase of heavy-isotope-labelled amino acid accumulation. Consumption rates were determined by linear regression of amino acid levels in the growth medium of K562 cells over time. n = 6 biologically independent samples for import and n = 5 for consumption. Data are mean ± s.e.m. d , Intracellular amino acid levels ( n = 7 biologically independent samples) and pool turnover rates of K562 cells growing in RPMI. Pool turnover rates were inferred by dividing amino acid import rates in c by intracellular levels. Data are mean ± s.e.m. e , Screen scores for K562 SLC7A5 CRISPRi/a. f , Amino acid import rates for K562 SLC7A5 and non-targeting control (NTC) CRISPRi in RPMI and in low leucine. Data represent the slope ± SE determined from the linear regression of n = 7 biologically independent samples. g , Cellular consumption rates for K562 SLC7A5 and NTC CRISPRi in RPMI. h , Consumption of leucine from low-leucine medium by K562 SLC7A5 and NTC CRISPRi cells. In g and h , the data represent the slope ± SE determined from the linear regression of n = 6 biologically independent samples. The shaded area represents 95% confidence interval. i , Intracellular amino acid levels of K562 SLC7A5 and NTC CRISPRi cells growing in RPMI or in low leucine ( n = 8 biologically independent samples; data are mean ± s.e.m.). Source numerical data are available in Source data .
Because transporter function can be influenced by the composition of the environment, we tested the effect of medium composition more broadly by assaying CRISPRi/a cells targeting the seven SLC7 genes in a medium we formulated to contain all amino acids and five additional known SLC7 family substrates (citrulline, ornithine, creatine, creatinine and carnitine) at levels found in human plasma (physiological amino acid/PAA-RPMI) 22 . In contrast with PAA-RPMI, RPMI contains many amino acids at 0.4× to 10× the levels found in human plasma and only trace amounts of alanine, cysteine and the five additional SLC7 substrates (Supplementary Table 2 ). However, in competition assays, we found that RPMI phenotypes were reproduced in PAA-RPMI (Pearson correlation coefficient 0.94), suggesting that differences in amino acid and other substrate levels between RPMI and human plasma minimally influence SLC7 family phenotypes.
In contrast to other SLC7 transporters, we found no phenotype associated with SLC7A8 (LAT2) KD or OE in RPMI or PAA-RPMI, despite expression comparable to SLC7A5. In addition, SLC7A8 mRNA levels induced by CRISPRa surpassed those of SLC7A5 induced by CRISPRa (Fig. 1b and Extended Data Fig. 3c ). While SLC7A8 is annotated as an importer for neutral amino acids such as Leu, Ile, His and Phe 12 , 36 , our data suggest that SLC7A8 does not import amino acids in K562 cells under conditions of amino acid limitation.
CRISPRi/a directly changes amino acid transport rates
We next asked whether screen hits reflect direct changes to transport of the limiting amino acid by the transporter targeted by CRISPRi/a. We quantified amino acid import by incubating K562 cells in medium containing 16 heavy-labelled amino acids and measuring intracellular isotope accumulation over time using gas chromatography–mass spectrometry (GC–MS) ( Methods , Fig. 2b and Extended Data Fig. 3d ). As many transporters function as exchangers, mediating both import and export of amino acids 10 , 12 , we also determined net transport across the plasma membrane by measuring amino acid consumption from the culture medium (Extended Data Fig. 3d ). We determined absolute import and net transport rates using an external standard curve for all 20 amino acids 23 (Fig. 2c and Extended Data Fig. 3e–g ).
We found that, while most amino acids were consumed from the medium, five (Asn, Asp, Glu, Gly and Pro) were secreted, consistent with previous findings in non-small cell lung cancer cells 3 , and our observation that removing these amino acids from RPMI did not impact proliferation (Figs. 1d and 2c ). For 9 out of the 11 amino acids consumed by K562 cells, import exceeded net transport by more than sevenfold, indicating high bidirectional flux (Extended Data Fig. 4a ). We also measured intracellular free amino acid levels and extrapolated free amino acid pool turnover rates by dividing import rates by intracellular levels. We found that most amino acids were turned over in minutes, further demonstrating high flux in cells (Fig. 2d ).
Using transport assays, we assessed the impact of SLC7A5 KD on amino acid transport rates. SLC7A5, required for tumour growth in many settings 38 , showed no proliferation defect in CRISPRi screens in RPMI but a strong defect in a medium low in His, Ile, Leu, Met, Phe, Trp, Tyr or Val (Fig. 2e ). We observed a large reduction in the import rates of these amino acids in either RPMI or low Leu (Fig. 2f ), whereas import of all other amino acids was unchanged (Extended Data Fig. 4b ). Consistent with an absence of proliferation phenotype for SLC7A5 KD in RPMI, overall amino acid consumption rates were unchanged (Fig. 2g ). Despite a strong reduction in neutral amino acid transport caused by SLC7A5 KD in RPMI, import rates still exceeded consumption rates (Fig. 2f,g ). Thus, a strong perturbation to transport rates does not necessarily result in a growth defect.
In low-Leu medium, however, Leu import rates fell below RPMI consumption rates in control cells and were even lower in SLC7A5 KD cells, consistent with the reduced proliferation imposed by the low-amino-acid medium and the strong additional growth defect imposed by KD of SLC7A5 (Fig. 2f,g ). Specifically, Leu consumption rates in low-Leu medium were 6-fold lower in control cells and 14-fold lower in SLC7A5 KD cells compared with Leu consumption in RPMI (Fig. 2h ). Moreover, reduced import of SLC7A5 substrates correlated with lower intracellular levels in both RPMI and low Leu (Fig. 2i and Extended Data Fig. 4c ). Of note, import of SLC7A5 substrates other than Leu was higher in low Leu than in RPMI (Fig. 2f and Extended Data Fig. 4d,e ), revealing competition among multiple substrates for import by a single transporter. Overall, these results show that SLC7A5 KD reduces import of its amino acid substrates, impacting cell proliferation when the import of a specific amino acid falls below normal consumption rates. We conclude that many of our CRISPRi/a screen phenotypes reflect direct changes to transport of the limiting amino acid by the targeted transporter gene.
SLC7A6 and SLC7A7 can scavenge cationic amino acids
While SLC7A1, SLC7A2 and SLC7A3 use membrane potential to drive import of Lys and Arg, the exchangers SLC7A6 and SLC7A7 are thought to export Arg and Lys in physiological conditions 12 , 36 . We characterized SLC7A6 and SLC7A7 phenotypes using transport assays (Figs. 2a and 3a ). Surprisingly, we found that SLC7A7 OE conferred an advantage in low Lys, and SLC7A6 OE conferred an advantage in low Lys and low Arg, suggesting a potential role for these proteins as net importers. We measured amino acid import in RPMI and observed that OE of SLC7A6 or SLC7A7 increased Lys and Arg import (Fig. 3b,c ). The increase in import for SLC7A7 OE was similar to that for SLC7A1 OE (four- to eightfold), the primary importer of these amino acids in K562 cells (Fig. 3b and Extended Data Fig. 5a ). In screening conditions, SLC7A6 OE increased Arg and Lys import, while SLC7A7 OE increased Lys, but not Arg, import, consistent with screen scores (Fig. 3c ). These results show that SLC7A6 and SLC7A7 are strong cationic amino acid importers and highlight differences in Arg affinity, with SLC7A7 emerging as the lower-affinity Arg transporter.

a , Scores obtained in CRISPRi/a screens in low amino acid and in RPMI. b , SLC7A1 CRISPRi/a and SLC7A7 CRISPRa specifically alter the import of arginine and lysine in K562 cells in RPMI. c , SLC7A7 CRISPRa increases the import of lysine when lysine is present in the medium at growth-limiting concentrations. SLC7A6 CRISPRa increases the import of arginine and lysine in all conditions tested. The arrows highlight the import of Arg and Lys in RPMI, import of Lys in low-Lys conditions and import of Arg in low-Arg conditions. d , SLC38A3 CRISPRa increases import of histidine and glutamine into K562 cells in RPMI. e , CRISPRa of SLC43A1 and SLC43A2 induces a proliferation defect in K562 over a range of conditions (RPMI with regular FBS, RPMI with dialyzed FBS (dFBS), and RPMI modified such that amino acids match human plasma levels (PAA-RPMI)). Data are the mean ± s.e.m. of two biological replicates each with six technical replicates. f , CRISPRa of SLC43A1 increases import of isoleucine and valine into K562 cells. g , CRISPRa of SLC43A1 decreases intracellular levels of large neutral amino acids in K562 cells. Levels were determined from import assays in f ( n = 7 biologically independent samples; data are mean ± s.e.m.). h , CRISPRa of SLC43A1 increases the export rate of valine from K562 cells cultured in RPMI. In b – d , f and h , the rates were determined from a linear regression of n = 6 biologically independent samples. The data represent the slope ± SE normalized to a non-targeting control (NTC). Source numerical data are available in Source data .
SLC38A3 is a high-affinity histidine transporter
SLC38A3 (SNAT3) can transport His and Asn but is primarily considered a Gln transporter, resulting in net import or export depending on environmental conditions 39 . However, we found that OE of SLC38A3 conferred a proliferation advantage in low His but not low Gln (Fig. 3a ). When we measured amino acid import in RPMI, we observed an increase for both His and Gln (Fig. 3d and Extended Data Fig. 5b ). These data suggest that SLC38A3 functions as a high-affinity His and low-affinity Gln transporter in K562 cells.
SLC43A1 is a net exporter of large neutral amino acids
In addition to SLC7A5 and SLC7A8, the L-type amino acid transporter (LAT) family includes SLC43A1 (LAT3) and SLC43A2 (LAT4), annotated as low-affinity facilitated diffusers of neutral amino acids Leu, Phe, Ile, Met and Val 10 , 40 , 41 . We found that SLC43A1/A2 OE reduced proliferation in RPMI, and SLC43A1 OE conferred resistance to low Val but hypersensitivity to low His, Leu and Phe, indicating varying affinities for structurally similar amino acids (Fig. 3a ). These phenotypes suggest that SLC43A1 and SLC43A2 function as net exporters of amino acids in most conditions. We confirmed proliferation defects in RPMI and other complete media and also observed that SLC43A1 OE increased Ile import by 1.6–2.4-fold and Val import by 2.6–4.4-fold with minimal changes for other amino acids, in RPMI, PAA-RPMI, low-Leu and low-Val media (Fig. 3e,f and Extended Data Fig. 5c–e ). Despite increased import, intracellular Ile and Val levels were substantially reduced in SLC43A1 OE cells across conditions, as were the intracellular levels of other neutral amino acids, which fell by at least 50% in all four media (Fig. 3g ). An exception to this trend was an increase in intracellular Val in low Val conditions, consistent with the screen resistance phenotype (Fig. 3a,g ). SLC43A2 OE caused similar changes to amino acid import and levels (Extended Data Fig. 5f,g ). To test the hypothesis that SLC43A1 and SLC43A2 act as net exporters of neutral amino acids, we measured amino acid export in RPMI and found that export of Val was specifically increased upon SLC43A1 OE (Fig. 3h ). Despite reduced intracellular levels, export of Leu, Ile and Phe in SLC43A1 OE was similar to control, suggesting a higher intrinsic export capacity (Extended Data Fig. 5h ). Overall these data show that SLC43A1 and SLC43A2 primarily facilitate neutral amino acid export but are capable of net importing Ile and Val in low Ile/Val conditions. These results illustrate the environment’s influence on transport and the phenotypic complexity arising from bidirectional transport, underscoring the importance of quantifying both import and export flux to determine net transporter activity.
Serotonin is an anti-ferroptosis endogenous antioxidant
CRISPRi/a screens in low amino acid conditions also reveal indirect effects, such as changes in the demand for the limiting amino acid or in the cell’s tolerance to the low amino acid environment. For instance, OE of the Gly transporter SLC6A9 caused hypersensitivity to low Ser, as Gly-to-Ser conversion depletes the one-carbon pool (Fig. 1h ) 42 . Also, OE of the Glu/Asp transporter SLC1A2 caused resistance to low Gln, as it reduced demand for exogenous Gln, consistent with previous observations (Extended Data Fig. 6a,b ) 35 , 43 , 44 .
In K562 cells, low cystine was unique because it caused cell death (Fig. 1d ), probably via glutathione depletion and consequent induction of ferroptosis 32 , 33 . We hypothesized that screens performed in low cystine would also yield hits related to ferroptosis sensitivity. SLC7A11, the primary cystine importer in animal cells, was one of the strongest hits (Fig. 4a ) 45 . Iron transporters exhibited robust phenotypes in the CRISPRi screen, consistent with the role of iron in enabling ferroptosis. KD of ABCC1, a major multidrug efflux pump, conferred resistance, probably by reducing intracellular glutathione efflux 46 , suggesting that ABCC1 OE may render cells susceptible to ferroptosis, a potentially druggable vulnerability (Fig. 4a ).

a , Scores obtained in transporter CRISPRi/a screens in K562 cells in low-cystine medium. b , SLC6A4 CRISPRa provides a growth advantage in low cystine dependent on SLC6A4 activity. Phenotype scores were determined in competition assays in low-cystine medium replicating screen conditions and represent the average log 2 FC between a specific and non-targeting control (NTC) sgRNA. Fluoxetine, 10 μM; serotonin, 1 μM; 5-OH Trp, 1 μM. n = 3 biologically independent samples. c , SLC6A4 CRISPRa provides resistance to K562 cells over a range of cystine levels. Cell viability was quantified in a luminescent assay after 48 h in low cystine. n = 4 biologically independent samples. d , Serotonin protects K562 cells from death in low cystine and expression of SLC6A4 increases protection. Assay as in c , and viability was normalized to the initial cell count (T0). Fluoxetine, 5 μM; serotonin: 1 (+) or 10 (++) μM; 5-OH Trp, 1 (+) or 10 (++) μM. n = 6 biologically independent samples. e , Serotonin protects K562 cells from death independent of GPX4 activity. Assay as in d . Ferrostatin-1, 0.1 μM; RSL3, 1 μM; erastin, 5 μM. f , Serotonin protects A375 cells from ferroptosis only at high concentration. Assay as in d . n = 4 biologically independent samples. g , Addition of serotonin (1 (+) or 10 (++) μM) reduces lipid peroxide levels in K562 cells induced for ferroptosis, dependent on SLC6A4 expression. Each data point represents the average of 20,000 cells determined by flow cytometry. n = 3 biologically independent samples. Peroxide levels were normalized to the ‘+Cys’ condition represented by the horizontal dashed line (normalized to a value of 1). h , Serotonin protects Caco-2 cells from ferroptosis dependent on SLC6A4 expression and independent of GPX4 activity. Assay as in d . n = 10 biologically independent samples. i , A cartoon illustrating the role of serotonin and SLC6A4 in suppressing ferroptosis. Boxed areas are from this study; core ferroptosis pathway components and ferroptosis-modulating molecules were adapted from others 33 , 61 . In d – f and h , P values were determined using two-tailed unpaired Student’s t -tests. In b – h , data are mean ± s.e.m. Source numerical data are available in Source data .
Unexpectedly, we found that overexpressing SLC6A4, the serotonin transporter, conferred resistance to low cystine (Fig. 4a ). SLC6A4 is poorly expressed in K562 cells whereas SLC7A11 is well expressed, and CRISPRa induced strong and specific OE (Extended Data Fig. 6c ). We validated the resistance of SLC6A4 and SLC7A11 OE in screen-like competition assays and observed that addition of fluoxetine, a selective serotonin reuptake inhibitor (SSRI) 47 , to block serotonin transport by SLC6A4, reduced the resistance of SLC6A4 OE by at least 50% while addition of exogenous serotonin increased resistance (Fig. 4b ). The same effects were observed in short-term viability assays in single cell lines (Fig. 4c,d ). Thus, transport of serotonin into cells by SLC6A4 seems to underlie ferroptosis resistance. While not part of the RPMI formula, serotonin can be generated via oxidative degradation of Trp 48 . Addition of 5-hydroxytryptophan (5-OH Trp), the metabolic intermediate in the conversion of Trp into serotonin, had no effect on viability, suggesting it was either not protective or not imported (Fig. 4b,d ). We hypothesize that SLC6A4 OE confers an advantage in low cystine by increasing the transport of low levels of serotonin present in the culture medium and consequent inhibition of ferroptosis.
To understand serotonin’s impact in the ferroptosis pathway, we tested its ability to mitigate ferroptosis in the presence of two inducers targeting different steps: erastin (SLC7A11 inhibitor), and RSL3 (glutathione peroxidase 4 (GPX4) inhibitor). Exposure of K562 cells to RSL3 alone or RSL3 plus erastin in low cystine did not prevent rescue by serotonin present in the medium or added exogenously, indicating serotonin acts downstream of GPX4 (Fig. 4e ). Similar results were obtained in the BRAF V600E melanoma cell line A375 (Fig. 4f ). Serotonin has affinity for unsaturated lipid bilayers and its indole ring has strong antioxidant activity 49 , 50 . We therefore measured lipid peroxide levels and found that serotonin almost completely abrogated the strong increase in peroxide levels caused by medium lacking cystine and containing RSL3; this effect was dependent on SLC6A4 OE (Fig. 4g and Extended Data Fig. 6d ). At high serotonin concentrations, rescue was independent of SLC6A4, probably due to other import mechanisms or passive diffusion.
K562 and A375 cells, which express SLC6A4 poorly, require SLC6A4 OE or high serotonin concentrations for anti-ferroptosis activity. We tested ferroptosis protection by serotonin via SLC6A4 in intestinal Caco-2 cells, which express high levels of SLC6A4 (Extended Data Fig. 6c ). In Caco-2 cells, serotonin addition to low-cystine medium prevented cell death and KD of SLC6A4 blocked rescue (Fig. 4h ). Rescue occurred downstream of GPX4, as demonstrated by RSL3 independence. Low-cystine conditions did not induce SLC6A4 expression in K562 and Caco-2 cells, in contrast to SLC7A11 (Extended Data Fig. 6e ). These data demonstrate that serotonin suppresses ferroptosis by quenching lipid peroxides and expression of SLC6A4 substantially potentiates its effect (Fig. 4i ).
Transporter essentiality is highly condition specific
We next assessed transporter essentiality using CRISPRi screening across a range of conditions in which growth was not deliberately nutrient limited. We hypothesized that condition-specific transporter phenotypes would not only shed light on transporter function but also provide insights into cellular metabolism and environmental effects, as transport flux is influenced by cellular demands and extracellular nutrient levels. First, CRISPRi screens in K562 cells cultured in complete RPMI revealed that about 9% of all KDs were significantly and reproducibly depleted (46 essential transporters) (Fig. 5a,b and Extended Data Fig. 7a–c ). We measured essentiality across three complete synthetic media and found that the majority of transporters exhibited condition-specific phenotypes (Fig. 5c ). For example, the folate transporter SLC19A1 was essential only in RPMI at low cell density or with dialysed foetal bovine serum (dFBS). Additionally, under high-density growth conditions, KD of the choline importer FLVCR1 (SLC49A1) and the bicarbonate transporter SLC4A7 resulted in strong defects while KD of the mitochondrial citrate transporter SLC25A1 conferred an advantage 17 , 28 . In contrast, a minority of KD phenotypes (for example, ABCE1, SLC25A10 and SLC39A9) were insensitive to changes in extracellular nutrient levels (Fig. 5c,d ).

a , Essential transporters in K562 cells growing in RPMI determined using CRISPRi screening. n = 2 screens each with two technical replicates. P values were determined using a Mann–Whitney test. b , A chord plot displaying all essential transporters from a . c , A tile plot of growth scores determined in CRISPRi screens in K562 cells across culture conditions ( n = 2 replicates). All transporters significantly enriched or depleted in at least one condition are included. ‘#’ highlights transporters discussed in the main text. d , A comparison of essentiality between conditions. Data represent growth scores for all transporters as determined in c . e , Growth complementation assays identifying SLC39A10 as the main zinc importer in K562 cells and manganese as a competing ion. Phenotypes were determined in RPMI + FBS, in RPMI + dFBS or in RPMI + dFBS supplemented with 10 μM metal ion. Data are mean ± s.e.m. of three technical replicates for non-targeting control (NTC) and three technical replicates of two biological replicates for SLC39A9 and SLC39A10. f , A cartoon illustrating the role of the mitochondrial pyruvate carrier (MPC1/MPC2) and the major sources and uses of cytoplasmic and mitochondrial pyruvate. Exogenous AKB was used to restore NAD + levels. ETC, electron transport chain. g , The addition of either alanine or of molecules restoring NAD + levels alleviates the defect of MPC1/2 CRISPRi. Phenotypes were determined in competition assays in RPMI + FBS and in RPMI + dFBS with 130 μM alanine, 1 mM AKB, 1 mM pyruvate, 4.5 mM lactate or 130 μM alanine + 4.5 mM lactate. Data are mean ± s.e.m. of two biological replicates each with three technical replicates. Horizontal dashed lines highlight the growth phenotype in the ‘+dFBS’ condition. h , A comparison of transporter essentiality in K562 and A375 cells. Data ( n = 2 replicates) are the phenotype scores determined in CRISPRi screens in RPMI for all transporters significantly depleted in at least one cell line. In a and d , black circles indicate transporter genes and red circles indicate negative control genes. In e and g , P values were determined using two-tailed unpaired Student’s t -tests. Source numerical data are available in Source data .
We next identified the nutrients causing condition-specific phenotypes in complementation assays. For example, comparing essentiality in cells cultured in RPMI with dFBS versus regular foetal bovine serum (FBS) revealed three transporters (SLC39A10, MPC1 and MPC2) with markedly different phenotypes (Fig. 5d ). SLC39A10 KD conferred a strong defect in dFBS but not in FBS, unlike KD of the closely related gene SLC39A9, which showed consistent deleterious effects in both conditions. SLC39 transporters are annotated as zinc importers, although some members import other metal ions such as cadmium and manganese 51 . Since dialysis removes small molecules and ions, and RPMI lacks transition metals, we hypothesized that SLC39A10 imports one or more essential metals for K562 cells. In contrast, SLC39A9 is probably buffered from extracellular metal ion concentration changes due to its localization in the Golgi 51 . We added specific metal ions to RPMI with dFBS and observed that zinc rescued the defect of SLC39A10 KD and manganese exacerbated it (Fig. 5e and Extended Data Fig. 7d ). These results suggest that SLC39A10 is a major zinc importer in K562 cells and that manganese can compete for import.
KD of MPC1 and MPC2, the two subunits of the mitochondrial pyruvate carrier (MPC) that transports cytoplasmic pyruvate into the mitochondria (Fig. 5f ), was also selectively detrimental in RPMI with dFBS. The defect of MPC KD in dFBS was rescued by addition of alanine (Fig. 5g and Extended Data Fig. 7e ), consistent with previous observations showing that MPC knockouts are deleterious in low-alanine conditions because mitochondrial pyruvate is required to synthesize alanine 52 . Lactate and pyruvate, present in FBS but removed by dialysis (Extended Data Fig. 7f ), displayed differential effects when added to MPC KD cells. Lactate exacerbated proliferation defects, whereas pyruvate alleviated them. In addition, supplementation with either pyruvate or alanine rescued defects caused by lactate (Fig. 5g and Extended Data Fig. 7g ). Given lactate and pyruvate’s impact on the NAD + /NADH ratio, we hypothesized that growth defects were caused by a reduction in the NAD + /NADH ratio 53 . Supplementing media with α-ketobutyrate (AKB), which increases the NAD + /NADH ratio without contribution to the tricarboxylic acid (TCA) cycle or to ATP generation 54 , rescued defects caused by MPC KD (Fig. 5g ). These results are consistent with mitochondrial pyruvate being required for alanine synthesis and NAD + regeneration.
We noted cell-type-specific transporter requirements by comparing transporter essentialities in K562 and A375 cells, in which CRISPRi/a is highly effective 55 . While many transporters showed similar essentiality between cell lines, 17 transporters exhibited strong cell-line-specific phenotypes (Fig. 5h ). For example, KD of the mitochondrial serine transporter SFXN1 (ref. 56 ) was deleterious in K562 cells but had no effect in A375 cells, despite the KD being similarly effective in the two cell lines (Extended Data Fig. 7h ). These findings suggest that K562 is unable to compensate for the loss of mitochondrial one-carbon metabolism by activating the cytosolic counterpart 57 and highlight the potential of comparing transporter essentiality to reveal cell-specific changes in metabolism.
Transporter CRISPRi/a screens in subcutaneous tumours
Given our findings that transporters are highly sensitive to the environment, we performed screens in xenografted tumours as a more physiological environment. We performed CRISPRi and CRISPRa transporter screens in K562 and A375 cells growing in subcutaneous tumours in immunodeficient mice. To ensure sufficient library coverage, we first determined engraftment efficiencies using green fluorescent protein (GFP)-labelled cells (Extended Data Fig. 8a,b ). Across all screens we identified transporter perturbations affecting proliferation in subcutaneous tumours (Fig. 6a,b ). To discern in vivo-specific effects, we screened in RPMI and also in human plasma-like medium (HPLM) 22 and adult bovine serum (ABS), which approximate blood nutrient levels 45 (Fig. 6c,d ). We quantified similarity across environments and found that CRISPRi phenotypes in HPLM and RPMI were strongly correlated (Pearson’s r > 0.9), and both of these had weaker but similar correlations to xenografts ( r = 0.7–0.8) (Fig. 6e,f ). CRISPRa correlations were generally lower, suggesting greater environmental dependency. In K562, xenograft phenotypes correlated more strongly with those in HPLM ( r = 0.7) than in RPMI ( r = 0.5), suggesting HPLM mimics the tumour environment better than RPMI 23 , and ABS scores poorly correlated with other conditions.

a , b , Pools of K562 and A375 transporter CRISPRi/a libraries were injected subcutaneously in the flank of immunodeficient mice. Growth scores were determined from enrichment/depletion of sgRNAs in whole tumour homogenates ( n = 4). The red circles represent negative control genes. c , d , A comparison of transporter CRISPRi/a phenotypes in subcutaneous (subQ) tumours to phenotypes in growth culture media identifies condition-specific effects. Pools of cells prepared in a and b were cultured in ABS (100% bovine serum), in HPLM and in RPMI. Data represent all transporters significantly enriched or depleted in at least one condition, and growth scores determined from two replicates were normalized to the most depleted transporter in each screen. ‘#’ highlights transporters discussed in the main text. e , f , Pearson correlation coefficients determined from pairwise comparison of growth scores for enrichments/depletions in c and d and P values were determined using a two-sided test. **: P < 1 × 10 −4 ; ***: P < 1 × 10 −6 . In a and b , P values were determined using a Mann–Whitney test. In a , c and e , data are from CRISPRi screens. In b , d and f , data are from CRISPRa screens. Source numerical data are available in Source data .
These results highlight nutrient dependencies and limitations in subcutaneous tumours and how they change across environments. For example, KD of the main glucose importer, SLC2A1, was deleterious in A375 cells in all tested conditions, while OE enhanced tumour growth. In contrast, SLC2A1 KD in K562 cells showed no phenotype, and OE was deleterious in tumours and RPMI. We observed comparable SLC2A1 expression levels at baseline in the two cell lines, and OE was similarly effective at increasing transcript (approximately tenfold) and plasma membrane protein levels (Extended Data Fig. 8c,d ), indicating that these differences probably stem from changes in glucose metabolism rather than in SLC2A1 activity, and a higher glucose demand in A375 cells is consistent with reports of increased glycolysis due to the BRAF V600E oncogene 58 . In a second example, the defects induced by either SLC6A9 OE, SLC19A1 KD or SFXN1 KD in K562 tumours suggest that maintaining the one-carbon pool is especially critical in the subcutaneous environment, as these transport perturbations all impair the folate cycle 56 .
Our findings in low-amino-acid media show that OE of nutrient transporters confers a growth advantage when the substrate is limiting proliferation. For instance, the advantage conferred by SLC2A1 OE in A375 tumours indicates that glucose is probably limiting proliferation of A375 cells in that environment (Fig. 6d ). Additionally, the advantage conferred by OE of amino acid transporters SLC7A3, SLC38A2 and SLC7A11 suggests that Arg, Gln and Cys levels are also limiting proliferation. Conversely, no growth advantage from transporter OE was observed in K562 tumours, suggesting a lack of nutrient limitations. This is consistent with the absence of proliferation defects upon KD of the major amino acid transporters (SLC1A5, SLC7A1, SLC7A5 and SLC7A11) in K562 tumours. Differences between A375 and K562 in nutrient limitations may reflect differences in demand or concentrations within the tumour environments, warranting further investigation.
Efforts are ongoing to identify essential nutrients for tumour growth and leverage this for improved anti-cancer therapy 59 . In this study, we developed a CRISPRi/a screening strategy to systematically identify nutrient transporters in cells, focusing on characterizing amino acid transport in the K562 leukaemia cell line. While major amino acid transporter families are well described 10 , 12 , our work enables the simultaneous interrogation of the contribution of each of the 64 annotated amino acid transporters to import and export across different conditions. To understand which transporters are essential or capable of import in nutrient-poor conditions, we performed screening at amino acid concentrations that limit proliferation. As these concentrations are generally lower than plasma or tissue interstitial fluid levels 22 , 23 , our growth-based screens probably identify high-affinity transporters, and not all the low-affinity transporters that also contribute to physiological amino acid homeostasis. We explored low-affinity transport mechanisms in individual CRISPRi/a cell lines in transport assays, and by performing screens in nutrient-rich and physiological conditions. Overall, we identified one or more SLCs required for the transport of 13 different amino acids, and complementary transporters able to import amino acids when overexpressed.
A notable finding is that amino acid transport involves high bidirectional import and export flux at the membrane, aligning with proposed models of cellular amino acid homeostasis 12 , 13 . While net transport typically favours amino acid import for cell proliferation, some transporters like SLC43A1 and SLC43A2 act as net exporters, only becoming importers when their substrates are limiting. This challenges assumptions about SLC43A1 as an importer and its pursuit as an anti-cancer drug target 41 . Similar phenotypes were observed for SLC16A10 (TAT1), indicating potential net exporter functions (Extended Data Fig. 2b ). These results emphasize the importance of measuring net flux across the membrane and considering the environment when assessing transport.
Our study shows that serotonin reduces lipid peroxidation, protecting cells from ferroptosis. Consistent with these findings, a recent study by Liu et al. also found that serotonin prevents ferroptosis by acting as a radical-trapping antioxidant 60 . Serotonin’s antioxidant effect requires transport across the plasma membrane by SLC6A4, highlighting that lipid peroxides causing ferroptosis are probably located in intracellular membranes 33 , 61 . Varying across human tissues, serotonin levels and SLC6A4 expression peak in the gut 62 , central nervous system or in diseases like carcinoid tumours 63 . Our results suggest that serotonin’s antioxidant role may be prominent in such tissues with elevated serotonin and SLC6A4 levels 64 . Importantly, inhibitors of SLC6A4, such as SSRIs, negate the protective effect of serotonin and might increase ferroptotic sensitivity in those environments 65 .
CRISPRi/a screening in subcutaneous xenografts identified nutrient dependencies and limitations in the TME. Although phenotypes in subcutaneous tumours were strongly correlated with in vitro conditions, certain transporters displayed large differences in essentiality between the in vivo and in vitro environments and further studies will be required to understand these effects. Although the TME is generally considered nutrient poor 2 , specific constraints on tumour growth remain unclear and have not been systematically explored. Our study, along with others 35 , 66 , demonstrates that identifying growth advantages from transporter OE highlights nutrients functionally limiting tumour growth. A375 melanoma cells faced glucose and amino acid limitation in xenograft tumours, while K562 cells showed no notable limitations, indicating potential variations in nutrient accessibility or environmental metabolite levels within tumours. These results align with observations that tissue interstitial fluid metabolite levels in lung cancer and pancreatic ductal adenocarcinoma xenografts are markedly different 23 .
In conclusion, our CRISPRi/a screening platform systematically queries transporter activity and function, providing insights into nutrient uptake and secretion under diverse growth conditions in vivo and in vitro. Beyond nutrients, this method is suitable for studying drug transport and the impact of metabolism on drug sensitivity, as most small-molecule drugs are actively imported into cells via SLCs and can be exported via ABC transporters 67 , 68 , 69 . We anticipate broader applications of our approach in exploring various aspects of transporter biology.
The research in this manuscript complies with relevant ethical regulations. The research was approved by Harvard University’s Committee on Microbiological Safety, and animal experiments were approved by the Massachusetts Institute of Technology Institutional Animal Care and Use Committee.
Cell culture and chemicals
Cell lines were from ATCC: K562 (CCL–243), A375 (CRL–1619), C2BBe1 clone of Caco-2 (CRL–2102) and HEK293T (CRL–3216). Cells used in this study were from low-passage cultures from primary stocks, and cell lines were not further authenticated. K562 cells were grown in RPMI (Corning 10–040) supplemented with 10% (v/v) heat-inactivated FBS (Gibco 10438026). HEK293T and A375 cells were grown in Dulbecco’s modified Eagle medium (DMEM, Corning 10–013) supplemented with 10% (v/v) FBS. Caco-2 cells were grown in Eagle’s minimal essential medium (EMEM, ATCC 30–2003) supplemented with 10% (v/v) FBS. All cell lines were grown at 37 °C and 5% CO 2 , and penicillin and streptomycin were added to all growth media to final concentrations of 100 U ml −1 and 100 μg ml −1 , respectively (Corning 30–002–CI). Cells were tested for mycoplasma contamination using the MycoAlert mycoplasma detection kit (Lonza LT07-318).
(1 S ,3 R )-RSL3 (Cayman Chemical 19288), erastin (MedChemExpress HY–15763) and ferrostatin-1 (Selleckchem S7243) were dissolved in dimethyl sulfoxide (DMSO), quality controlled and stored by the ICCB-Longwood screening facility. Fluoxetine HCl (Millipore Sigma F132), serotonin HCl (Tocris 354750) and 5-OH Trp (Millipore Sigma H9772) were prepared in DMSO. For metal ion complementation assays, ZnSO 4 ·7H 2 O (Fluka 96500), CuSO 4 (VWR VW 3312–2), Fe(NO 3 ) 3 ·9H 2 O (Sigma F–1143) and MnCl 2 ·4H 2 O (Sigma M8530) were dissolved in H 2 O to 100 mM. Lactic acid (Fluka 69775) was adjusted to pH 7 with NaOH and diluted with H 2 O to 0.5 M, and sodium pyruvate (Sigma P2256) and AKB (Sigma K401) were dissolved in H 2 O to 300 mM and 1 M, respectively.
Transporter CRISPRi/a sgRNA library cloning
Transporter libraries include sgRNAs targeting 413 SLC genes as defined by the Human Genome Organization gene nomenclature, 28 atypical SLCs and 48 ABC transporters (45 as defined by the Human Genome Organization and 3 atypical). sgRNA sequences were from Horlbeck et al. 34 and included 10 sgRNAs per gene for all genes, except 37 of them that had 2 transcription start sites and were therefore represented by 20 sgRNAs, and 730 NTC sgRNAs (Supplementary Table 1 ). The design and cloning of sgRNAs was performed as previously reported 34 with minor modifications. A single pool of oligonucleotides (12,000) for both CRISPRi and CRISPRa libraries was synthesized by Twist Biosciences. PCR reactions were set up using primers specific to either CRISPRi or CRISPRa sequences using Phusion polymerase (NEB) according to the manufacturer’s instructions and using three different conditions to minimize amplification bias (HF buffer, GC buffer and GC buffer + 3% DMSO). Each 50 μl reaction included 4.5 ng template and 0.4 μM primers, and was amplified using eight cycles of 30 s at 98 °C, 30 s at 60 °C (57 °C for the DMSO-containing reaction) and 10 s at 70 °C preceded by 1 min at 98 °C and followed by 1 min at 72 °C. Two replicate sets of reactions were run, and all CRISPRi and CRISPRa amplified libraries were pooled separately. The amplified library (88 bp) was purified by agarose gel electrophoresis and extracted using the QIAquick gel extraction kit (Qiagen). Purified PCR products were digested with BlpI and BstXI, and the digested fragment was purified on a 20% polyacrylamide Tris–borate–EDTA (TBE) gel (Thermo Fisher Scientific). Digested fragments were purified by isopropanol precipitation, and DNA concentrations were quantified by fluorescence using the Qubit dsDNA high-sensitivity assay kit (Thermo Fisher Scientific). Plasmid pU6-sgRNA EF1Alpha-puro-T2A-BFP (Addgene #60955) was digested using BlpI and BstXI, purified by agarose gel electrophoresis and extracted using the QIAquick gel extraction kit (Qiagen). Digested PCR fragments were ligated into the restricted vector at a 1:1 molar ratio using T4 DNA ligase and were purified using the MiniElute PCR purification kit (Qiagen). Purified ligated plasmids were electroporated into MegaX DH10B (Thermo Fisher Scientific) and plated on LB/Amp plates. Library coverage was 10,000× for both libraries and was determined by serial dilution and colony counting. After 15 h at 30 °C, the lawn of bacteria was scraped off the plates, and plasmid DNA was prepared using the Plasmid Plus Maxi kit (Qiagen).
Lentivirus preparation
HEK293T cells were transfected with the lentiviral plasmid, psPAX2 (Addgene #12260) and pCMV-VSV-G (Addgene #8454) in a 2:2:1 molar ratio using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. The growth medium was replaced 6 h post-transfection and was then collected at 24–30 h and 48–54 h post-transfection. The two collected growth medium fractions were pooled, centrifuged at 1,000 g for 10 min and filtered through a 0.45 μm low-protein-binding membrane. Lentivirus-containing supernatants were stored at −80 °C.
Media preparation
To prepare complete RPMI lacking all amino acids, 8.59 g of RPMI without amino acids, sodium phosphate (US Biological R8999–04 A), 2.00 g sodium bicarbonate (Sigma S6014) and 0.80 g sodium phosphate dibasic (Sigma S0876) were diluted in 945 ml de-ionized H 2 O. After addition of 100 ml dFBS (Gibco, Thermo Fisher Scientific 26400044) and 10 ml of penicillin–streptomycin 100× solution (Corning 30–002–CI) and homogenization, the medium was sterilized via 0.22 μm membrane filtration (RPMI + dFBS without amino acids). Amino acids were added to this base medium as needed from stock solutions to RPMI levels or to low amino acid screen concentrations, as described in Supplementary Tables 2 and 3 . To make complete RPMI used as the control arm in low amino acid screens and experiments, all 19 amino acids were added to complete RPMI without amino acids to RPMI levels (RPMI rich).
Complete RPMI with amino acids present at physiological levels (PAA-RPMI) was prepared from RPMI + dFBS without amino acids as above, except that 965 ml de-ionized H 2 O was used. To that, amino acids were added to their concentration in human plasma 22 using the same stock solutions as for RPMI above. In addition to the amino acids present in RPMI, alanine, cysteine, carnitine, citrulline, creatine, creatinine, N -acetylglycine and ornithine were also added to their level in human plasma. Quantities added are described in Supplementary Table 2 . For low amino acid conditions, the corresponding amino acid was added such that the final concentration in PAA-RPMI matched the concentration used in the CRISPRi/a screens.
To prepare RPMI medium for amino acid import assays (RPMI with 16 amino acids present as heavy isotopes), we first added unlabelled cystine, OH-Pro, Met and Trp to RPMI + dFBS without amino acids. This base medium consisting of complete RPMI lacking 16 amino acids was then used to prepare heavy-labelled RPMI and RPMI with low Leu or low Val. Stock solutions of heavy-labelled amino acids (Cambridge Isotope Laboratories) were added to RPMI lacking 16 amino acids to their concentration in RPMI or to the screen concentration (for Leu and Val), as outlined in Supplementary Table 2 .
To prepare PAA-RPMI for amino acid import assays, we prepared a complete PAA-RPMI base as above but lacking 16 amino acids. We then added heavy-labelled amino acids from the same stocks as for RPMI to the concentration in PAA-RPMI, as outlined in Supplementary Table 2 .
RPMI + FBS and DMEM + FBS were prepared as outlined in ‘Cell culture and chemicals’ section. HPLM (Thermo A4899101) was adjusted with 10% dFBS. ABS (Sigma-Aldrich B9433) thawed at 4 °C was adjusted to 50 μM with cystine·2HCl (Sigma C6727). After 1 h at 37 °C to ensure dissolution, ABS was filtered through a 0.2 μm low-protein-binding membrane.
Amino acid titrations
Single amino acid dropout RPMI medium was prepared for each of the 19 amino acids present in RPMI by adding 18 amino acids to complete RPMI without amino acids. K562 cells grown in complete RPMI were washed 3× using cycles of centrifugation for 5 min at 300 g and resuspension of the cell pellet in phosphate-buffered saline (PBS, Corning 21–040 CV). The final cell pellet was first resuspended in PBS at a density of 10 million ml −1 and then added to single amino acid dropout RPMI or complete RPMI to a final density of 0.1 million ml −1 . Thirty microlitres of these cell suspensions was pipetted into the wells of 384-well microplates (Thermo Fisher Scientific 164610). Amino acid stock solutions in H 2 O (as described in Supplementary Table 2 ) were adjusted to 0.005% Triton X-100 using a 100× stock solution in H 2 O and were added to wells of the microplates using a D300 digital dispenser (Hewlett-Packard). We confirmed that the highest concentration of Triton X-100 (0.33 parts per million) did not affect proliferation of K562 cells. Each amino acid was tested along a twofold dilution series from 1× to 1/1,024× its concentration in RPMI and including a no amino acid control. Wells on plate edges were filled but not used for any measurements. The drug dispensing arrangement for each amino acid was spatially randomized (and re-organized during data analysis) to minimize bias. Each data point was present in quadruplicates, and three separate identical plates were prepared for viability measurement after 24, 48 and 72 h incubation. Assay plates were incubated at 37 °C/5% CO 2 inside containers humidified by sterile wet gauze. After 24, 48 or 72 h, plates were removed from incubation and cooled at room temperature for 10 min, before dispensing 30 μl of CellTiter-Glo (CTG) (Promega) (1:1 dilution in PBS) into each well. Following a 10 min incubation at room temperature, luminescence was measured in a plate reader (BioTek Synergy H1). A control plate with K562 cells dispensed in complete RPMI was prepared and luminescence was monitored at the onset of the experiment ( T = 0) and after 24, 48 and 72 h incubation in the same conditions to determine K562 doubling rates in RPMI. Each data point was averaged across four replicates, divided by luminescence at T = 0, was internally normalized within each plate and was displayed on a log 2 scale to represent the total number of population doublings.
Preparation of CRISPRi/a parental cell lines
To generate a K562 cell line stably expressing dCas9-KRAB (K562 CRISPRi), K562 cells were transduced with lentiviral particles produced using vector pMH0001 (Addgene #85969; which expresses dCas9-BFP-KRAB from a spleen focus forming virus promoter with an upstream ubiquitous chromatin opening element) in the presence of 8 mg ml −1 polybrene (Sigma). A pure polyclonal population of dCas9-KRAB-expressing cells was generated by two rounds of fluorescence-activated cell sorting (FACS) gated on the top half of BFP-positive cells (BD FACS Aria II) (Extended Data Fig. 8e ). In a third round of FACS sorting, single cells from the top half of BFP-positive cells were sorted into microplates to establish monoclonal cell lines. The performance of K562 CRISPRi monoclonal lines in knocking down endogenous genes was evaluated by individually targeting three control genes (ST3GAL4, SEL1L and DPH1) and measuring gene expression changes by RT–qPCR, and the best-performing monoclonal cell line was selected for all work described in this study. The preparation of monoclonal K562 cells expressing CRISPRa machinery (K562 CRISPRa) and of A375 expressing CRISPRi (polyclonal) and CRISPRa (monoclonal) has been described elsewhere 25 , 55 .
Preparation of CRISPRi/a cell lines using sgRNAs targeting individual genes and expression analysis by RT–qPCR
Pairs of complementary synthetic oligonucleotides (Integrated DNA Technologies) forming sgRNA protospacers flanked by BstXI and BlpI restriction sites were annealed and ligated into BstXI/BlpI double-digested plasmid pU6-sgRNA EF1Alpha-puro-T2A-BFP (Addgene #60955). Oligonucleotides used to build sgRNAs targeting individual genes are listed in Supplementary Table 4 . The sequence of all sgRNA expression vectors was confirmed by Sanger sequencing, and lentiviral particles were produced using these vectors as described above (‘Lentivirus preparation’ section). Parental CRISPRi/a cells were infected with individual sgRNA expression vectors by addition of lentivirus supernatant to the culture medium in the presence of 8 μg ml −1 polybrene. Transduced cells were selected using puromycin (2 μg ml −1 for K562 cells) starting 48 h post-transduction and over the course of 7 days with daily addition of the antibiotic. After 24 h growth in puromycin-free medium, 0.1 million cells were collected and total RNA was extracted using the RNeasy Plus Mini kit (Qiagen). Complementary DNA was synthesized from 0.1–0.5 μg total RNA using Superscript IV reverse transcriptase (Invitrogen) and oligo(dT) 20 primers (Invitrogen) following the manufacturer’s instructions. Reactions were diluted two- to fivefold with H 2 O, and qPCR was performed using PowerUp SYBR Green PCR Master mix (Thermo Fisher Scientific), 2 μl diluted cDNA preparation and 0.4 μM of primers using a QuantStudio 6 Real-Time PCR system (Thermo Fisher Scientific). All qPCR primers are listed in Supplementary Table 4 . The expression level of target genes was determined relative to the housekeeping gene GAPDH (log 2 fold change (FC) versus GAPDH) by subtracting Ct values (ΔCt). Cells were grown in RPMI or in low amino acid medium for 72 h at low confluence with daily media changes before expression profiling.
For Caco-2 cells, we used a one-vector CRISPRi system (Addgene #71236) to create polyclonal KD cell lines. Pairs of complementary synthetic oligonucleotides (Integrated DNA Technologies) forming sgRNA protospacers flanked by BsmBI restriction sites were annealed and ligated into BsmBI digested plasmid pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro. Caco-2 cells were infected using lentiviral supernatant produced from these plasmids. Transduced cells were selected using EMEM containing puromycin at 2–5 μg ml −1 over 12 days. Changes in expression level due to CRISPRi were quantified as above. Cells were grown in RPMI or in low-cystine medium for 72 h at low confluence with daily media changes before expression profiling.
Transporter CRISPRi/a screens in low amino acid medium
Lentiviral supernatant was prepared for both the transporter CRISPRi and CRISPRa sgRNA libraries as described above (‘Lentivirus preparation’ section) and was stored at −80 °C. The multiplicity of infection (MOI) of both preparations was determined by titration onto target cell lines and quantification of the percentage of BFP + cells 2–3 days post-transduction by flow cytometry (BD Biosciences LSR II).
For transporter CRISPRi/a screens in low amino acid, K562 CRISPRi (or CRISPRa) parental cells (85–100 million) were transduced with lentiviral supernatant at an MOI of 0.25–0.3 in 250 ml culture medium + 8 μg ml −1 polybrene in a 225 cm 2 cell culture flask (Costar). Twenty-four hours post-transduction, cells were collected and resuspended in 200 ml fresh medium in 2 × 225 cm 2 flasks. Starting 48 h post-transduction, the culture medium was exchanged daily and cells were maintained at 0.5–1.0 million ml −1 in puromycin (1.75 μg ml −1 ) in 400–500 ml. After 6 days in puromycin, the proportion of BFP + cells determined by flow cytometry increased to 94–96% of the fraction of viable cells. After recovery for 1 day in puromycin-free medium, 2 × 10 million library cells were collected and stored at −80 °C ( T = 0 samples). The remaining cells were grown for 24 h in RPMI/rich (complete RPMI with all amino acids; ‘Media preparation’ section). A total of 120 million cells were collected by centrifugation at 300 g for 5 min and washed twice with 25 ml PBS, and the final cell pellet was resuspended in 4.8 ml PBS. Complete RPMI medium with 18 amino acids present at RPMI level and 1 amino acid present at a concentration that limits growth of K562 (‘Media preparation’ section) was prepared, and 50 ml was added to 150 cm 2 tissue culture flasks (Falcon 355001) that were preheated and equilibrated to 37 °C/5% CO 2 . Library cells were added to the flasks to a final density of 0.1 million ml −1 . For all amino acids except Trp and Cys, screens were conducted over the course of 16 days with three cycles of 1 day in RPMI/rich and 4 days in RPMI/low amino acid, followed by a final day in RPMI/rich. The growth medium was changed daily on all flasks using centrifugation of a fraction of the culture and resuspension of the pellet into pre-equilibrated growth medium. For medium change after growth in RPMI/rich, cell pellets were washed twice with PBS before resuspension in low-amino-acid medium. The amount of culture centrifuged was adjusted such that the final cell density after medium exchange was 0.1 million ml −1 . For Trp and Cys, screens were conducted over the course of 16 days and medium was changed every 36 h (1× 24 h RPMI/rich; 4× pulses of 36 h RPMI/low amino acid; 1× 36 h RPMI/rich; 4× pulses of 36 h RPMI/low amino acid; 1× 36 h RPMI/rich). The RPMI/rich control arm and the low His, low Arg and low Lys were run in technical replicates. At the end of the screen, 12–15 million cells were collected by centrifugation, washed once with PBS and stored at −80 °C. The number of population doublings for each screen is indicated in Supplementary Table 3 . Genomic DNA (gDNA) was extracted from cell pellets using the QIAamp DNA Blood Mini Kit according to the manufacturer’s instruction, except that the elution was performed using 10 mM Tris HCl pH 8.5. Typical yields from 15 million cells ranged from 90 to 240 μg gDNA. sgRNA barcodes were amplified by PCR using the gDNA from at least 6 million cells as template and Phusion (NEB M0530) as polymerase. An equimolar mix of primers with stagger regions of different length (CC_LSP_025 to CC_LSP_032_c) was used as forward primer, and barcoded index primers (CC_Cri_a_rev1 to CC_Cri_a_rev10) were used as reverse primers. Reactions were composed of 1× HF buffer, 0.2 mM dNTPs, 0.4 μM forward primer mix, 0.4 μM indexed reverse primer, 0.5 μl Phusion, 1.5 mM MgCl 2 and 5 μg gDNA in a volume of 50 μl. After 30 s at 98 °C, the reactions were subjected to 23 cycles of 98 °C for 30 s, 62 °C for 30 s and 72 °C for 30 s, and were followed by 72 °C for 5 min. All reactions from each screen were pooled, and the amplified PCR product (~240–250 bp) was purified by agarose gel electrophoresis using the QIAquick gel extraction kit (Qiagen). Purified PCR products were quantified by fluorescence using the Qubit dsDNA high-sensitivity assay kit (Thermo Fisher Scientific). Individual indexed libraries were mixed in equimolar ratio and were further purified using a QIAquick PCR purification kit (Qiagen). After quantification by qPCR using the NEBnext library quant kit for Illumina (NEB), pooled libraries were sequenced on an Illumina HiSeq 2500 platform using a 50 bp single read on a high-output standard v4 flow cell with a 10–20% PhiX spike-in. About 15 million reads were obtained for each indexed screen.
For some of the screens, an alternative strategy was used to accommodate sequencing on an Illumina NextSeq 500 platform. Changes to the protocol above include amplification of extracted gDNA by PCR using a barcoded forward primer (CC_fwd1 to CC_fwd20) and a barcoded reverse primer (CC_Cri_a_rev1 to CC_Cri_a_rev20) using Q5 polymerase (NEB M0491L). Pooled libraries were sequenced on an Illumina NextSeq 500 platform using a 75 bp single read on a high-output flow cell with a 2–5% PhiX spike-in. A total of 15–30 million reads were obtained for each indexed screen.
Sequencing data were analysed as previously reported 25 with the following modifications. Trimmed sequences were aligned to the library of protospacers present in the transporter CRISPRi/a sgRNA libraries (Supplementary Table 1 ), and 89–92% of the number of raw reads typically aligned to the library of protospacers. To estimate technical noise in the screen, simulated negative control genes (the same number as that of real genes) were generated by randomly grouping 10 sgRNAs from the pool of 730 NTC sgRNAs present in the libraries. For each gene (and simulated control gene), which is targeted by ten sgRNAs, two metrics were calculated: (1) the mean of the strongest 7 rho phenotypes by absolute value (‘phenotype score’) and (2) the P value of all 10 rho phenotypes compared with the 730 NTC sgRNAs (Mann–Whitney test). To display data compactly, we calculated a single score (‘screen score’) by multiplication of the phenotype score with −log 10 ( P value). sgRNAs were required to have a minimum of 100 counts in at least one of the two conditions tested to be included in the analysis. To deal with the noise associated with potential low count numbers, a pseudocount of 10 was added to all counts. Gene-level phenotype scores and P values are available in Supplementary Tables 5 and 6 .
For screens in low amino acid conditions, scores were calculated using two technical replicates for Arg, Lys and Val. For all other conditions, scores were calculated using one replicate for the low amino acid condition and two technical replicates for both T0 and untreated samples. The highest-scoring negative control gene in any condition was used as the significance cut-off for all conditions (for CRISPRa, a significant hit had a screen score >0.425 or <−0.45; for CRISPRi, >0.49 or <−0.40). We excluded significant hits that had single scores <−0.12 in the untreated control arm of the screen unless they were either hypersensitive or very strong resistant (>1.5) hits in any of the low amino acid conditions. This exclusion was performed because a slowdown in proliferation rate leads to pan-resistance across screen conditions, as previously reported 25 .
For in vivo screens, scores were determined without normalization to the number of population doublings. The number of sgRNAs required in at least one of two conditions tested was lowered to 25. For comparison with in vitro screens, screen scores were normalized to the most hypersensitive hit (set to −1) in a given screen. Significance cut-offs were determined by the highest-scoring pseudo negative gene (K562 CRISPRi, <−0.075 and >0.025; K562 CRISPRa, <−0.15 (−0.20 for subcutaneous) and >0.05 (0.10 for subcutaneous and ABS); A375 CRISPRi, <−0.05 and >0.03 (0.06 for subcutaneous); A375 CRISPRa, <−0.17 (−0.25 for subcutaneous) and >0.1 (0.7 for subcutaneous).
Phenotype similarity across environments was determined using all genes with a significant deleterious or beneficial phenotype in any of the environments tested. Pairwise Pearson correlations (and P values) were calculated using the function rcorr in the R package Hmisc (v. 4.7.1), and data were displayed using the R package corrplot (v. 0.84).
For CRISPRi screens in K562 in RPMI, two biological replicates each consisting of two technical replicates were collected. To compute a volcano plot of the data, the read counts of the two technical replicates were averaged after normalization to the total number of reads and the resulting two replicates were processed in the analysis pipeline, as explained above. Essential transporters were determined by averaging screen scores determined for each biological replicate. A stringent significance threshold was determined by the highest-scoring pseudogene in any of the two replicates before averaging (score <−0.13). Essential transporters were arranged in a chord plot using GOPlot (v. 1.0.2).
For K562 CRISPRi screens in different culture conditions, significance cut-offs for each screen were determined by the highest-scoring pseudogene. Genes that were significant in at least one condition were displayed in tile plots. However, genes that did not have a growth score <−0.20 or >0.10 in any condition were excluded from the plots. For the comparison between K562 and A375 transporter essentiality, phenotype scores were used instead of growth scores to account for potential differences in sgRNA efficacy. Genes having a phenotype score <−0.065 were significant and were displayed in the tile plot.
Transporter CRISPRi/a screens in rich medium
For K562 CRISPRi/a transporter screens in non-growth-limited conditions, pooled libraries were prepared as above. Cells were collected by centrifugation at 300 g for 5 min, washed in PBS and resuspended in PBS at 100 million ml −1 . Screens were initiated by addition of 7.5 million cells into 50 ml medium in 150 cm 2 flasks and were conducted by passaging the libraries in growth medium for 14 days with medium exchange at least each 2 days and keeping the density between 0.1 million ml −1 and 0.4 million ml −1 . The pools of cells were collected at T = 14 days and T = 0 days, and enrichment was analysed as outlined above. For RPMI + 10% FBS at high cell density, cells were kept at a density of 0.3–1.2 million ml −1 with medium exchange every 2 days.
For A375 screens, CRISPRi/a parental cells grown in RPMI + 10% FBS in 4× 15 cm cell culture dishes (Falcon 353025) to 80–90% confluence were transduced with transporter library lentiviral supernatant at an MOI of 0.25–0.3 in presence of 8 μg ml −1 polybrene. Starting 48 h post-transduction, cells were passaged daily to maintain <95% confluence in RPMI + 10% FBS + puromycin (1.0 μg ml −1 ). After 4 days in puromycin, the proportion of BFP + cells determined by flow cytometry increased to 90–95% of the fraction of viable cells. After recovery for 1 day in puromycin-free medium, library cells were trypsinized and then quenched. Cells were collected by centrifugation at 300 g for 5 min, washed in PBS and resuspended in PBS at 100 million ml −1 . Screens were initiated by addition of 15 million cells into 50 ml medium in 15 cm dishes and were conducted by passaging the libraries in growth medium for 14 days with medium exchange at least every 2 days and keeping the confluence between 20% and 80%. Cells were collected at T = 14 days and T = 0 days, and enrichment was analysed as outlined above.
Transporter screens in subcutaneous tumours in immunodeficient mice
All animal experiments conducted in this study were approved by the MIT Institutional Animal Care and Use Committee. A maximum tumour burden of 2 cm was permitted per Institutional Animal Care and Use Committee protocol, and these limits were not exceeded. Male mice between 3 and 4 months old were used in this study. All animals were housed at ambient temperature and humidity (18–23 °C, 40–60% humidity) with a 12 h light and 12 h dark cycle and co-housed with littermates with ad libitum access to water. For animal injections, pooled cells were washed twice with PBS and filtered through a 40 μm cell strainer. After centrifugation at 300 g for 5 min, pelleted cells were resuspended at 100 million ml −1 in PBS and the suspensions were kept on ice until injection (<1 h). NOD.Cg- Prkdc scid Il2rg tm1Wjl /SzJ mice (NSG mice; The Jackson Laboratory, strain #005557) were injected subcutaneously into both flanks with 0.1 ml of pooled cell suspension (10 million cells per tumour). Each screen was conducted with three mice ( N = 6 tumours) over 14 days. At the endpoint, animals were killed and whole tumours (typically 100–500 mg) were excised and kept at −80 °C. gDNA was extracted from homogenized whole tumours (at least 160 μg for each tumour) using the DNeasy Tissue kit (Qiagen). sgRNA barcodes were amplified by PCR using 160 μg of gDNA, as described above for transporter screens in low amino acid screens. Libraries were quantified, pooled and analysed as described earlier. For each screen, the two samples that had the greatest number of NTC counts not within 1 log 2 of the median were excluded from the analysis to reduce technical noise. Remaining replicates were paired and read counts were averaged after correcting to match total read counts. These averaged samples were then processed with the T = 0 days samples to determine gene-level scores as outlined above.
Determination of engraftment frequency in subcutaneous tumours in NSG mice
Lentivirus was prepared from plasmid pLJM1–eGFP (Addgene #19319). K562 CRISPRi + transporter library cells were infected with pLJM1–eGFP lentivirus, and the proportion of GFP + cells was determined by flow cytometry 72 h post-transduction. GFP + cells were spiked into K562 CRISPRi + transporter library to a final ratio of 1 to 1 × 10 3 , 1 to 1 × 10 4 or 1 to 1 × 10 5 . For each dilution and unspiked control, cells were prepared as above, diluted with PBS, and 100 μl (0.1, 1 or 10 million cells) was injected into both flanks of NSG mice. In addition, a portion of each preparation was collected for T = 0 days samples and for growth in RPMI + FBS. Tumours were allowed to form over the course of 19 (10 million cells injected), 26 (1 million) and 34 days (0.1 million). At the endpoint, animals were killed and whole tumours were excised and kept on ice. Tumours were dissociated, and the presence of GFP + cells was determined by flow cytometry analysis. To determine the skewness of the NTC sgRNA distribution, tumours were analysed in the same way as the screens.
Immunoblotting
RPMI medium containing low amino acid was made in the same way as for CRISPRi/a screens. In addition, RPMI containing no Arg, His or Lys was prepared analogously, and we used RPMI/rich and RPMI without amino acids as controls. For the time course, K562 cells growing in RPMI/rich were collected, washed twice with treatment medium, and resuspended in treatment medium at 0.35–0.45 million ml −1 . After 24 h, cells were pelleted and resuspended in fresh medium at 0.35–0.45 million ml −1 . For each timepoint, 0.6 million K562 cells were pelleted by centrifugation at 300 g for 3 min and flash frozen in liquid nitrogen. Pellets were lysed in 1× RIPA buffer (50 mM Tris HCl, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate and 1% NP40) and lysates clarified by centrifugation at 21,000 g for 10 min at 4 °C. Protein concentration in lysates was quantified by bicinchoninic acid assay (Pierce). Then, 20–25 μg protein was run on a 4–20% Bis-Tris Bolt gel (NuPAGE) in 1× MES buffer (NuPAGE) at 125 V for 90 min, and transferred to 0.45 μm nitrocellulose at 18 V for 60 min using the Trans-Blot SD semi-dry transfer system (Bio-Rad) in Tris–glycine transfer buffer (10 mM Tris base, 0.1 M glycine and 20% MeOH). The membrane was blocked with 5% skimmed milk or bovine serum albumin (for phospho-antibodies) in TBST for 1 h at room temperature, and incubated overnight at 4 °C with primary antibody in the same solution used for blocking. After 3× 5 min washes with TBST, incubation for 1–3 h at room temperature with 1:5,000 secondary antibody (Cell Signaling Technology goat HRP-linked anti-rabbit IgG #7074), and three more 5 min washes with TBST, blots were developed in 2 ml Western Lighting Plus chemiluminescent substrate (PerkinElmer) and imaged on the ImageQuant LAS4000 (Cytiva). After imaging, the blot was incubated for 15 min in Restore Western Blot Stripping Buffer (Thermo Fisher Scientific) followed by 1 h in 30% H 2 O 2 at 37 °C. After extensive washes with TBST, the blot was blocked and reprobed as above. For each experiment, a single blot was serially assayed with the following rabbit antibodies (Cell Signaling Technology): vinculin (E1E9V) #13901 dilution 1:2,000; phospho-p70 S6 kinase (Thr389) #9234 dilution 1:1,000; phospho-eIF2α (Ser51) (D9G8) #3398 dilution 1:1,000; phospho-4E-BP1 (Ser65) #9451 dilution 1:1,000; p70 S6 kinase #9202 dilution 1:1,000; eIF2α #9722 dilution 1:2,000; 4E-BP1 (53H11) #9644 dilution 1:2,000.
For plasma membrane fractions, we prepared a fresh 10 mM solution of EZ-link-sulfo-NHS-SS-biotin reagent (Thermo Fisher Scientific A39258) in H 2 O. Cells were grown in low amino acid media for 72 h at low confluence with daily media changes or in regular RPMI. Cells (5–20 million) were washed twice with ice-cold PBS and resuspended to 40 million ml −1 in PBS. EZ-link-sulfo-NHS-SS-biotin was added to 800 μM, and cells were incubated for 30 min at room temperature with gentle rocking. Cells were pelleted and resuspended in 500 μl 150 mM glycine and incubated for 2 min. After washing with 1 ml PBS, cell pellets were resuspended in lysis buffer (50 mM Tris HCl pH 7.5, 100 mM NaCl, 5 mM EDTA pH 8, 1% Triton X-100, 1× protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific 1861280)) at 22.2 μl per 1 million cells. Cells were lysed at 4 °C for 10 min, and lysates were clarified by centrifugation at 21,000 g for 10 min at 4 °C. Supernatants (‘total extracts’; about 3.6 μg μl −1 ) were stored at −80 °C. For western blots, 1/6 volume of 6× Laemmli sample buffer was added and samples were incubated at 95 °C for 5 min. For pull-downs, 100–300 μl total protein extract was added to a tube containing 25 μl of streptavidin agarose resin (Thermo Fisher Scientific 20357) washed once with lysis buffer. After rotating samples at 4 °C for 60 min, the resin was washed with 3× 500 μl lysis buffer and centrifugation at 500 g for 3 min. Biotinylated proteins were eluted by incubating washed beads with 1× Laemmli sample buffer (1 μl per 7.5 μg input total protein) at 95 °C for 5 min.
For plasma membrane fraction western blotting, 10–30 μg total protein extract and the equivalent of 30–90 μg input protein of pull-down samples were run on 4–15% Mini Protean TGX precast gels (Bio-Rad) and transferred to nitrocellulose using the iBlot3 system and reagents (Invitrogen) at 25 V for 3 min. Membranes were blocked in Intercept (TBS) Blocking Buffer (LI-COR) for 1–2 h at room temperature, and then incubated overnight at 4 °C with primary antibody in blocking buffer + 0.2 % Tween-20 (SLC7A5 (Proteintech, 28670-1-AP) 1:5,000; SLC3A2/4F2hc (Cell Signaling, D603P, #13180) 1:1,000; SLC7A6 (Novus Biologicals, NBP2-75086) 1:500; SLC7A7 (Novus Biologicals, NBP1-82826) 1:500; SLC2A1 (Cell Signaling, D3J3A, #12939) 1:1,000). After 3× 5 min washes with TBST, incubation for 1–3 h at room temperature with 1:20,000 anti-rabbit secondary antibody (IRDye800CW LI-COR 926-32211) or 1:10,000 anti-goat secondary antibody for SLC7A6 only (donkey IgG H&L, Alexa Fluor 750 conjugate, Abcam ab175744) in blocking buffer + 0.2% Tween-20 + 0.01% sodium dodecyl sulfate. After 3× 5 min washes with TBST, blots were imaged on the iBright FL1500 Imaging System (Invitrogen). For vinculin staining post-imaging, blots were re-processed as above starting with an overnight incubation in 1:2,000 vinculin (Cell Signaling, E1E9V, #13901) in blocking buffer + 0.2 % Tween-20.
Growth competition assays
Growth phenotypes were measured in competition assays where a test cell line was mixed with its corresponding NTC control at a 1:1 ratio. Cell lines were grown in complete RPMI, and cell densities were determined using a TC20 automated cell counter (Bio-Rad). Cell mixes were prepared by combining 1 million test cells with 1 million NTC cells, washing twice with 10 ml PBS, and resuspending the final pellet in 1 ml PBS. Fifty microlitres of resuspended cells (0.1 million) was added to wells of a 12-well plate containing 1 ml of growth medium, and an aliquot was stored at −80 °C to determine the initial ratio for each mix. Cultures were passaged as necessary (that is, daily for low amino acid screen validation) by centrifugation of a portion of the culture and resuspension in fresh medium. At the end of the experiment, the total number of population doublings was determined for all conditions and 0.1–0.2 million cells were collected by centrifugation. gDNA was extracted from cell pellets using the QIAamp DNA Blood Mini Kit according to the manufacturer’s instruction, except that the elution was performed using 10 mM Tris HCl pH 8.5 (typical yields were 2–10 μg). Each extracted gDNA sample was assessed in two qPCR reactions using either a forward primer complementary to the test cell sgRNA sequence or to the NTC sgRNA sequence, and a universal reverse primer. Primers were tested beforehand to ensure linearity and specificity of detection. The composition of the mix was estimated by the difference in Ct value between the two reactions (ΔCt). Growth phenotypes were determined by comparing the ΔCt post-growth in the test medium with the ΔCt of the sample taken at T = 0 (ΔΔCt), and then normalizing by the number of population doubling differences between the test medium and a vehicle control (for example, low amino acid versus RPMI/rich). To determine growth phenotypes in complete media, ΔΔCt values were normalized by the number of population doublings between T = 0 and the end of the assay.
Amino acid transport assays in K562 cells
The polymer coverslip in a 35 mm µ-Dish (Ibidi 81156) was coated with Cell-Tak. For each dish, 12.25 μg Cell-Tak (Corning 354240) diluted to 360 μl with H 2 O was neutralized with 40 μl 1 M bicarbonate pH8 solution and rapidly spread over the surface of the coverslip. After 45 min at room temperature, the solution was removed and the coverslip was thoroughly washed with 2× 800 μl H 2 O and subsequently left to dry for a few minutes. To prepare cells for immobilization, K562 were collected (1 million cells per dish), washed once with RPMI without FBS and resuspended in RPMI without FBS (400 μl per dish). Resuspended cells were added to coated dishes and incubated at room temperature for 30–45 min. Medium and unattached cells (typically, 0.8 million cells are needed to form a homogeneous monolayer of K562 cells) were gently removed by aspiration. After one wash with 500 μl RPMI/rich, cells were incubated for at least 2 h at 37 °C/5% CO 2 in 500 μl RPMI/rich to reach steady state.
For import assays, one dish is required for each timepoint for each cell line. We typically assayed amino acid import over seven to eight timepoints (0, 5, 10, 20, 30, 40, 100 and 250 s) to capture the initial slope for all 16 amino acids. RPMI/rich was removed by aspiration and was replaced by 400 μl pre-warmed and pre-equilibrated RPMI + 16 heavy-labelled amino acids. After incubation at 37 °C for the duration of the timepoint (on a heat block for short timepoints and in the incubator for longer timepoints), cells were thoroughly washed by sequentially submerging dishes into 4× 400–600 ml ice-cold PBS. For the 0 s timepoint, the same procedure as above was performed but using RPMI/rich instead of heavy-labelled medium and incubating for 10 s. PBS in the washes was refreshed regularly to ensure limited contamination of intracellular amino acid pools. After the last wash, PBS was removed by aspiration and 600 μl of ice-cold extraction buffer (80% MeOH/20% H 2 O spiked with 4 μg ml −1 norvaline (Sigma N7627)) was added. Cells were scraped off the plate on ice (United Biosystems MCS–200) and were transferred to 1.5 ml tubes. Samples were homogenized in a thermomixer at 2,000 rpm and 4 °C for 15 min. Tubes were spun at 21,100 g for 10 min at 4 °C, and supernatants were transferred to new tubes containing 10 μl of 0.1 N HCl and were stored at −80 °C until analysis by GC–MS.
For amino acid consumption assays, a single dish was required for all timepoints for each cell line. We typically assayed consumption over five timepoints (0, 1, 2, 3 and 4 h) in RPMI and over four timepoints (0, 5, 15 and 60 min) in low amino acid medium. RPMI/rich was removed by aspiration and was replaced by 300 μl pre-warmed and pre-equilibrated RPMI/rich. At each timepoint, 15 μl of medium was removed and kept on ice. All samples were spun at 1,000 g for 5 min at 4 °C, and 10 μl of the supernatant was transferred to a tube containing 600 μl of extraction buffer + 10 μl of 0.1 N HCl. Samples were stored at −80 °C until analysis by GC–MS.
For GC–MS analysis, samples were dried at room temperature using a flow of nitrogen. To each tube, we added 24 μl methoxamine reagent (ThermoFisher TS–45950) and incubated the resuspended samples at 37 °C for 1 h. We next added 30 μl N -methyl- N -( tert -butyldimethylsilyl)trifluoroacetamide + 1% tert -butyldimethylchlorosilane (Sigma 375934) and incubated homogenized samples at 80 °C for 2 h. After centrifugation at 21,100 g for 10 min, supernatants were transferred to glass vials. Derivatized samples were analysed on a DB-35MS column (Agilent Technologies) in an Agilent 7890B gas chromatograph linked to an Agilent 5977B mass spectrometer. One microlitre of sample was injected at 280 °C and mixed with helium carrier gas at a flow rate of 1.2 ml min −1 . After injection, the oven was held at 100 °C for 1 min and ramped to 250 °C at 3.5 °C min −1 . The oven was then ramped to 320 °C at 20 °C min −1 and held for 3 min at 320 °C. Electron impact ionization in the mass spectrometer was performed at 70 eV, and the MS source and quadrupole was held at 230 °C and 150 °C, respectively. Scanning mode was used for detection with a scanned ion range of 100–650 m / z . Acetone washes were performed after every three to five samples and before and after each run. GC–MS raw data were quantified using El-MAVEN (v0.11.0), and, for each amino acid, we extracted the area of the peak for the fragment with the highest signal over noise for the parent ion and all detectable isotopologues (fragments are highlighted in Fig. 2b ). Natural isotope abundance was subsequently corrected using IsoCorrectoR to determine total ion counts for each amino acid fragment 70 . For import assays, each sample was first normalized using the norvaline internal standard. We corrected each sample by a factor determined by calculating the ratio of the norvaline signal to the average of the norvaline signal over all timepoints. As the experiment was run under steady state conditions, we applied a second correction that took advantage of the fact that the total ion count for all amino acid fragments (labelled and unlabelled) remains constant over all timepoints. We calculated the total ion count (excluding norvaline) for each sample and corrected each sample by a factor determined by calculating the ratio of the total ion count for the sample to the average total ion count over all timepoints. To determine absolute amounts in each sample, we used a standard mix (Cambridge Isotope Laboratories MSK–A2) containing 17 amino acids to which we added Gln and Asn. We made dilutions of this mix in 0.1 N HCl and added them to 600 μl K562 cell extract prepared as for the import assay. We analysed these samples as above and applied a linear regression to the data. We then used the linear regression data and the mean norvaline signals of the standard and the sample to convert ion counts into pmol amino acid. Data were normalized by the number of cells used in the assay (0.8 million in cell monolayer minus an estimated 20% cell loss during the washing steps). Intracellular amino acid levels were calculated by averaging the sum of unlabelled and heavy-labelled amino acid over all the timepoints. To calculate import rates, we determined the initial slope of the increase in intracellular levels of heavy-labelled amino acids. We applied a linear regression to heavy-labelled amino acid levels over time, and the number of data points included was variable depending on the amino acid and the conditions. We typically used data from 0 to 100 s for Asn, Asp, Gln, Glu and Pro; data from 0 to 40 s for Arg, Gly, His, Ile, Lys, Ser, Thr and Val; data from 0 to 20 s for Leu and Tyr; and data from 0 to 10 s for Phe.
For import assays in low-arginine and low-lysine conditions (and RPMI control), we used liquid chromatography–mass spectrometry (LC–MS) to quantify light- and heavy-labelled amino acids in methanol cell extracts prepared in the same way as for GC–MS. Samples were dried down using a flow of nitrogen, resuspended in 100 μl 80% MeOH/20% H 2 O and filtered through a 10 kDa PES filter. LC–MS analysis was performed using a QExactive orbitrap mass spectrometer using an Ion Max source and heated electro-spray ionization probe coupled to a Dionex Ultimate 3000 UPLC system (Thermofisher). For this, 2.5 μl sample was injected onto a SeQuant ZIC-pHILIC 2.1 mm × 150 mm (5 μm particle size) column (Millipore Sigma). The flow rate was set to 0.15 ml min −1 , and temperatures were set to 30 °C for the column compartment and 4 °C for the autosampler tray. The following conditions were used to achieve chromatographic separation: mobile phase A was 20 mM ammonium carbonate, 0.1% ammonium hydroxide, and mobile phase B was 100% acetonitrile. The chromatographic gradient was as follows: 0–20 min, linear gradient from 80% to 20% B; 20–20.5 min, linear gradient from 20% to 80% B; 20.5–28 min, hold at 80% B. The mass spectrometer was operated in full scan, polarity-switching mode, the spray voltage was set to 4.2 kV and the heated capillary was held at 320 °C. The MS data acquisition was performed in a range of 70–1,000 m / z , with the resolution set at 70,000, the AGC target at 1 × 10 6 and the maximum injection time at 20 ms. Quantification of light- and heavy-labelled amino acid levels from LC–MS raw data and import rates were determined as for GC–MS without natural abundance correction and calculation of absolute levels.
For consumption experiments, each sample was normalized using the norvaline standard as above. We next adjusted sample signals to account for the decrease in volume of medium at each timepoint. Finally, we made use of the fact that the experiment was run under steady-state conditions to correct samples on the basis of the total ion count (excluding norvaline) of each sample. We applied a linear regression to the total ion count over the time course of each experiment. We then corrected each sample by a factor calculated by taking the ratio of the total ion count of the sample to the computed level determined by the regression. To determine absolute amounts in each sample, we diluted 10 μl of the standard mix prepared above into 600 μl extraction buffer + 10 μl RPMI/rich and analysed these standards as detailed for the samples above. We applied a linear regression to the standard data and converted sample ion counts to pmol using the regression data as well as the mean norvaline signals of the standards and samples. Consumption rates were calculated from the slope of the changes in amino acid levels over the time course of the assay determined by linear regression.
Amino acid export was measured by exposing cells to RPMI containing heavy-isotope-labelled amino acids, rapidly switching to regular RPMI and measuring the release of labelled amino acids into the medium. While this approach was technically challenging due to the rapidity of export and the effects of differences in intracellular amino acid levels on export rates, we were able to quantify export rates for a subset of amino acids. K562 cells attached in a monolayer on the surface of an Ibidi dish were prepared as for the import assay. Cells were incubated for at least 2 h with RPMI containing heavy-labelled amino acids at 37 °C/5% CO 2 . Growth medium was removed by aspiration, and cells were thoroughly washed by sequentially submerging dishes into 4× 400–600 ml PBS at room temperature. After aspiration of the last PBS wash, 160 μl pre-warmed and pre-equilibrated RPMI/rich was added to the dish. Plates were kept at 37 °C, and the medium was homogenized by regularly swirling the dishes. Samples of the media (12.5 μl) were taken over the course of the assay (0, 5, 10, 20, 30, 40 and 100 s) and kept on ice. Samples were centrifuged at 300 g for 5 min at 4 °C, and 10 μl of the supernatant was transferred to a tube containing 600 μl of extraction buffer + 10 μl of 0.1 N HCl. Samples were stored at −80 °C until analysis by GC–MS that was performed and analysed in the same way as the consumption samples.
Cell viability assays
For K562, cells growing in RPMI/10% FBS were collected by centrifugation, washed 2× with PBS and resuspended at 10 million ml −1 in PBS. Medium was prepared for each growth condition by addition of small molecules to RPMI low cystine using DMSO stocks. K562 cells were added to each medium to a final concentration of 0.1 million ml −1 , and suspensions were dispensed into wells of a 48-well plate in triplicates. After 72 h incubation at 37 °C/5% CO 2 , 50 μl of each culture was transferred to a 96-well plate (in duplicates), 50 μl CTG reagent was added and luminescence was measured in a plate reader (BioTek Synergy H1). Viability was quantified by normalization to luminescence at T = 0 h. For the cystine titration, K562 cells washed in PBS were resuspended in RPMI −Cys (0.1 million ml −1 ) and dispensed in a 96-well plate. Cystine was added using a D300 digital drug dispenser (HP), and viability at 72 h was determined by addition of CTG reagent and measurement of luminescence.
For A375, cells were trypsinized and resuspended in RPMI −Cys, then washed 2× with RPMI −Cys and finally resuspended at 10 million ml −1 in the same medium. Cells were added to growth medium to 0.1 million ml −1 and dispensed into wells of a 96-well plate. After 2 h at 37 °C/5% CO 2 , small molecules were added to wells using a D300 digital drug dispenser (HP) using stocks in DMSO. Viability was determined at T = 48 h by addition of CTG reagent and measurement of luminescence.
For Caco-2 assays, 96-well microplates were coated with poly- d -lysine to avoid cell loss during washing steps. Forty microlitres of a 1:1 (v/v) solution of poly- d -lysine (Sigma A–003–M) 0.1 mg ml −1 in H 2 O and PBS was added to each well and incubated for 1 h at room temperature. Wells were washed 3× with PBS and left to air dry. Caco-2 cells were seeded into the 96-well plates in EMEM/10% FBS at a density of about 1,000 cells per well. After 48 h at 37 °C/5% CO 2 , wells were washed twice with RPMI −Cys followed by addition of 50 μl RPMI −Cys. Small molecules were added to wells using a D300 digital drug dispenser (HP). Viability was quantified at T = 48 h using the CTG luminescence assay and compared with viability at T = 0 h.
Lipid peroxidation quantification
K562 cell lines growing in RPMI/rich were collected, washed 2× with PBS and incubated for 24 h in RPMI −Cys at 1 million ml −1 . After media exchange to fresh RPMI −Cys and dilution to 0.5 million ml −1 , cells were dispensed in a 12-well plate (2 ml per well). Cystine was added from a 200× aqueous stock solution, and RSL3 and serotonin were added using 1,000× stocks in DMSO. After 6 h incubation at 37 °C/5% CO 2 , 100 μl of a solution of BODIPY 581/591 C11 (Thermo Fisher D3861) 200 μM in RPMI −Cys (5% v/v DMSO) was added to each well. After 30 min incubation at 37 °C/5% CO 2 , cells were washed once with PBS and analysed by flow cytometry (BD LSRFortessa). The oxidized form of BODIPY C11 was quantified using 488 nm emission and 530/30 nm emission, and the reduced form was quantified using 561 nm emission and 610/20 nm emission. Peroxide levels were estimated by the ratio of these two fluorescence intensities.
Statistics and reproducibility
All experimental assays, excluding CRISPR screens, were performed at least in triplicate. The exact number of replicates is indicated in the figure legends. In general, data represent the mean of individual replicates and error bars represent the standard error of the mean (s.e.m.). Data analysis and display was accomplished in R (v. 3.6.0) using ggplot (v. 3.3.3). Significance probabilities, except for CRISPR screens, were calculated using two-tailed unpaired Student’s t -tests. For western blots, experiments were reproduced twice with similar results and one of the replicates was shown as representative example. No sample size calculation was performed for experiments in cell culture. For in vivo screens, we pre-determined in initial experiments that sufficient library representation was achieved in every single tumour and we included six replicate tumours for each screen condition. To improve data quality in these screens, we excluded from each set of screens the two datasets with the highest amount of technical noise. Noise was determined in an unbiased way by quantifying the number of NTC sgRNA counts that were within 1 log 2 of the median after growth in vivo. No other data were excluded from the study. Transporter CRISPR screens were run in duplicates, and all attempts at replication were successful (Extended Data Fig. 2c,d ). Mice were randomly chosen for the four subcutaneous injection conditions. Other experiments were not randomized as there was no allocation into different experimental groups. The researcher performing subcutaneous injections was blinded to the identity of the cells injected. All pooled CRISPR screens are inherently randomized and blinded. Other experiments were not blinded as all data were collected using unbiased methods.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All CRISPR screening data are provided in Supplementary Tables 5 and 6 . The composition of the sgRNA libraries is provided in Supplementary Table 1 . Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding authors on reasonable request.
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12 , 31–46 (2022).
Article CAS PubMed Google Scholar
Pavlova, N. N. & Thompson, C. B. The emerging hallmarks of cancer metabolism. Cell Metab. 23 , 27–47 (2016).
Article CAS PubMed PubMed Central Google Scholar
Hosios, A. M. et al. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev. Cell 36 , 540–549 (2016).
Lomelino, C. L., Andring, J. T., McKenna, R. & Kilberg, M. S. Asparagine synthetase: function, structure, and role in disease. J. Biol. Chem. 292 , 19952–19958 (2017).
Choi, B.-H. et al. Lineage-specific silencing of PSAT1 induces serine auxotrophy and sensitivity to dietary serine starvation in luminal breast tumors. Cell Rep. 38 , 110278 (2022).
Delage, B. et al. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 126 , 2762–2772 (2010).
Ngo, B. et al. Limited environmental serine and glycine confer brain metastasis sensitivity to PHGDH inhibition. Cancer Discov. 10 , 1352–1373 (2020).
Bröer, S. Amino acid transporters as targets for cancer therapy: why, where, when, and how. Int. J. Mol. Sci. 21 , 6156 (2020).
Article PubMed PubMed Central Google Scholar
Meixner, E. et al. A substrate-based ontology for human solute carriers. Mol. Syst. Biol. 16 , e9652 (2020).
Pizzagalli, M. D., Bensimon, A. & Superti-Furga, G. A guide to plasma membrane solute carrier proteins. FEBS J. 288 , 2784–2835 (2021).
Dvorak, V. et al. An overview of cell-based assay platforms for the solute carrier family of transporters. Front. Pharm. 12 , 722889 (2021).
Article CAS Google Scholar
Bröer, S. & Bröer, A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem. J. 474 , 1935–1963 (2017).
Article PubMed Google Scholar
Gauthier-Coles, G. et al. Quantitative modelling of amino acid transport and homeostasis in mammalian cells. Nat. Commun. 12 , 5282 (2021).
Girardi, E. et al. Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat. Commun. 11 , 6145 (2020).
Kory, N. et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci. Adv. 6 , eabe5310 (2020).
Luongo, T. S. et al. SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature 588 , 174–179 (2020).
Kenny, T. C. et al. Integrative genetic analysis identifies FLVCR1 as a plasma-membrane choline transporter in mammals. Cell Metab. 35 , 1057–1071.e12 (2023).
Wang, Y. et al. SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. Nature 599 , 136–140 (2021).
Shi, X. et al. Combinatorial GxGxE CRISPR screen identifies SLC25A39 in mitochondrial glutathione transport linking iron homeostasis to OXPHOS. Nat. Commun. 13 , 2483 (2022).
César-Razquin, A. et al. A call for systematic research on solute carriers. Cell 162 , 478–487 (2015).
Kampmann, M. CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem. Biol. 13 , 406–416 (2018).
Cantor, J. R. et al. Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell 169 , 258–272.e17 (2017).
Sullivan, M. R. et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8 , e44235 (2019).
Jost, M. et al. Combined CRISPRi/a-based chemical genetic screens reveal that rigosertib is a microtubule-destabilizing agent. Mol. Cell 68 , 210–223.e6 (2017).
Palmer, A. C., Chidley, C. & Sorger, P. K. A curative combination cancer therapy achieves high fractional cell killing through low cross-resistance and drug additivity. eLife 8 , e50036 (2019).
Tian, R. et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat. Neurosci. 24 , 1020–1034 (2021).
Allikmets, R., Gerrard, B., Hutchinson, A. & Dean, M. Characterization of the human ABC superfamily: isolation and mapping of 21 new genes using the expressed sequence tags database. Hum. Mol. Genet . 5 , 1649–1655 (1996).
Hediger, M. A., Clémençon, B., Burrier, R. E. & Bruford, E. A. The ABCs of membrane transporters in health and disease (SLC series): introduction. Mol. Asp. Med. 34 , 95–107 (2013).
Schuetz, J. D. et al. MRP4: a previously unidentified factor in resistance to nucleoside-based antiviral drugs. Nat. Med. 5 , 1048–1051 (1999).
Nasr, R. et al. Molecular analysis of the massive GSH transport mechanism mediated by the human Multidrug Resistant Protein 1/ABCC1. Sci. Rep. 10 , 7616 (2020).
Maddocks, O. D. K. et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493 , 542–546 (2013).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149 , 1060–1072 (2012).
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22 , 266–282 (2021).
Horlbeck, M. A. et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5 , e19760 (2016).
Rebsamen, M. et al. Gain-of-function genetic screens in human cells identify SLC transporters overcoming environmental nutrient restrictions. Life Sci. Alliance https://doi.org/10.26508/lsa.202201404 (2022).
Fotiadis, D., Kanai, Y. & Palacín, M. The SLC3 and SLC7 families of amino acid transporters. Mol. Asp. Med 34 , 139–158 (2013).
To, T.-L. et al. A compendium of genetic modifiers of mitochondrial dysfunction reveals intra-organelle buffering. Cell 179 , 1222–1238.e17 (2019).
Kanai, Y. Amino acid transporter LAT1 (SLC7A5) as a molecular target for cancer diagnosis and therapeutics. Pharmacol. Ther. 230 , 107964 (2022).
Rubio-Aliaga, I. & Wagner, C. A. Regulation and function of the SLC38A3/SNAT3 glutamine transporter. Channels 10 , 440–452 (2016).
Babu, E. et al. Identification of a novel system L amino acid transporter structurally distinct from heterodimeric amino acid transporters. J. Biol. Chem. 278 , 43838–43845 (2003).
Wang, Q. & Holst, J. L-type amino acid transport and cancer: targeting the mTORC1 pathway to inhibit neoplasia. Am. J. Cancer Res. 5 , 1281–1294 (2015).
PubMed PubMed Central Google Scholar
Labuschagne, C. F., van den Broek, N. J. F., Mackay, G. M., Vousden, K. H. & Maddocks, O. D. K. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 7 , 1248–1258 (2014).
Alkan, H. F. et al. Cytosolic aspartate availability determines cell survival when glutamine is limiting. Cell Metab. 28 , 706–720.e6 (2018).
Tajan, M. et al. A role for p53 in the adaptation to glutamine starvation through the expression of SLC1A3. Cell Metab. 28 , 721–736.e6 (2018).
Muir, A. et al. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. eLife 6 , e27713 (2017).
Cao, J. Y. et al. A genome-wide haploid genetic screen identifies regulators of glutathione abundance and ferroptosis sensitivity. Cell Rep. 26 , 1544–1556.e8 (2019).
Lin, L., Yee, S. W., Kim, R. B. & Giacomini, K. M. SLC transporters as therapeutic targets: emerging opportunities. Nat. Rev. Drug Discov. 14 , 543–560 (2015).
Bellmaine, S., Schnellbaecher, A. & Zimmer, A. Reactivity and degradation products of tryptophan in solution and proteins. Free Radic. Biol. Med. 160 , 696–718 (2020).
Azouzi, S. et al. Antioxidant and membrane binding properties of serotonin protect lipids from oxidation. Biophys. J. 112 , 1863–1873 (2017).
Peters, G. H. et al. Binding of serotonin to lipid membranes. J. Am. Chem. Soc. 135 , 2164–2171 (2013).
Jeong, J. & Eide, D. The SLC39 family of zinc transporters. Mol. Asp. Med. 34 , 612–619 (2013).
Rossiter, N. J. et al. CRISPR screens in physiologic medium reveal conditionally essential genes in human cells. Cell Metab. 33 , 1248–1263.e9 (2021).
Luengo, A. et al. Increased demand for NAD + relative to ATP drives aerobic glycolysis. Mol. Cell 81 , 691–707.e6 (2021).
Sullivan, L. B. et al. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162 , 552–563 (2015).
Gerosa, L. et al. Receptor-driven ERK pulses reconfigure MAPK signaling and enable persistence of drug-adapted BRAF-mutant melanoma cells. Cell Syst. 11 , 478–494.e9 (2020).
Kory, N. et al. SFXN1 is a mitochondrial serine transporter required for one-carbon metabolism. Science 362 , eaat9528 (2018).
Ducker, G. S. et al. Reversal of cytosolic one-carbon flux compensates for loss of mitochondrial folate pathway. Cell Metab. 23 , 1140–1153 (2016).
Yun, J. et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325 , 1555 (2009).
Luengo, A., Gui, D. Y. & Vander Heiden, M. G. Targeting metabolism for cancer therapy. Cell Chem. Biol. 24 , 1161–1180 (2017).
Liu, D. et al. Tryptophan metabolism acts as a new anti-ferroptotic pathway to mediate tumor growth. Adv. Sci. 10 , 2204006 (2023).
Lei, G., Zhuang, L. & Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 22 , 381–396 (2022).
Bertrand, P. P., Bertrand, R. L., Camello, P. J. & Pozo, M. J. Simultaneous measurement of serotonin and melatonin from the intestine of old mice: the effects of daily melatonin supplementation. J. Pineal Res. 49 , 23–34 (2010).
Halperin, D. M. et al. The frequency of carcinoid syndrome at neuroendocrine tumor diagnosis: a large population-based study using SEER-Medicare data. Lancet Oncol. 18 , 525–534 (2017).
Berger, M., Gray, J. A. & Roth, B. L. The expanded biology of serotonin. Annu. Rev. Med. 60 , 355–366 (2009).
Ryan, S. K. et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat. Neurosci. 26 , 12–26 (2023).
Unlu, G. et al. Metabolic-scale gene activation screens identify SLCO2B1 as a heme transporter that enhances cellular iron availability. Mol. Cell 82 , 2832–2843.e7 (2022).
Nigam, S. K. What do drug transporters really do? Nat. Rev. Drug Discov. 14 , 29–44 (2015).
Dobson, P. D. & Kell, D. B. Carrier-mediated cellular uptake of pharmaceutical drugs: an exception or the rule? Nat. Rev. Drug Discov. 7 , 205–220 (2008).
Girardi, E. et al. A widespread role for SLC transmembrane transporters in resistance to cytotoxic drugs. Nat. Chem. Biol. 16 , 469–478 (2020).
Heinrich, P. et al. Correcting for natural isotope abundance and tracer impurity in MS-, MS/MS- and high-resolution-multiple-tracer-data from stable isotope labeling experiments with IsoCorrectoR. Sci. Rep. 8 , 17910 (2018).
Nicholson, B., Sawamura, T., Masaki, T. & MacLeod, C. L. Increased Cat3-mediated cationic amino acid transport functionally compensates in Cat1 knockout cell lines. J. Biol. Chem. 273 , 14663–14666 (1998).
El-Brolosy, M. A. et al. Genetic compensation triggered by mutant mRNA degradation. Nature 568 , 193–197 (2019).
Wang, T., Wei, J. J., Sabatini, D. M. & Lander, E. S. Genetic screens in human cells using the CRISPR–Cas9 system. Science 343 , 80–84 (2014).
Download references
Acknowledgements
We thank A. Muir, N. Matheson, Z. Li, S. Block and the Sorger and Vander Heiden labs for discussions, and J. Weissman, D. Trono, B. Weinberg, D. Sabatini and C. Gersbach for plasmids. This work was supported by NIH/NCI grant U54-CA225088 to P.K.S., grants R35-CA242379 and P30-CA014051 to M.G.V.H., a Jane Coffin Childs Memorial Fund for Medical Research fellowship to A.M.D., Ludwig Cancer Research, the Termeer and Lustgarten Foundations, and the MIT Center for Precision Cancer Medicine.
Author information
Authors and affiliations.
Laboratory of Systems Pharmacology, Harvard Medical School, Boston, MA, USA
Christopher Chidley, Benjamin L. Gaudio & Peter K. Sorger
Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, USA
Alicia M. Darnell, Evan C. Lien, Anna M. Barbeau & Matthew G. Vander Heiden
Department of Biology, Massachusetts Institute of Technology, Cambridge, MA, USA
Anna M. Barbeau & Matthew G. Vander Heiden
Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA
Matthew G. Vander Heiden
Department of Systems Biology, Harvard Medical School, Boston, MA, USA
Peter K. Sorger
You can also search for this author in PubMed Google Scholar
Contributions
C.C. designed the study and performed experiments. C.C. analysed data with help from A.M.D. C.C. and A.M.D. performed cellular transport assays and western blots. B.L.G. assisted with experiments. E.C.L., A.M.B. and C.C. performed mouse screens. M.G.V.H. and P.K.S. supervised the project and obtained funding. C.C. and P.K.S. wrote the paper. A.M.D. edited the paper.
Corresponding authors
Correspondence to Christopher Chidley or Peter K. Sorger .
Ethics declarations
Competing interests.
M.G.V.H. is a scientific advisor for Agios Pharmaceuticals, iTeos Therapeutics, Sage Therapeutics, Auron Therapeutics and Droia Ventures. P.K.S. is a co-founder and member of the BOD of Glencoe Software, a member of the BOD of Applied Biomath and a member of the SAB of RareCyte, NanoString and Montai Health, and a consultant for Merck. None of these relationships has influenced the content of this manuscript. The other authors declare no competing interests.
Peer review
Peer review information.
Nature Cell Biology thanks Stefan Broer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended data fig. 1 transporter crispri/a screening optimization..
( a ) Composition of the CRISPRi/a transporter sgRNA libraries. ( b ) Preparation of K562 CRISPRi or CRISPRa pooled transporter libraries. Custom sgRNA libraries for CRISPRi and CRISPRa were constructed and packaged into lentivirus. A K562 monoclonal cell line expressing either the CRISPRi or CRISPRa machinery was infected with lentiviral particles, and untransduced cells were removed by antibiotic selection. Because transporter expression is often tissue-specific 20 and cell lines can lose transporter expression over time 71 , CRISPRa bypasses the need for transporter expression. CRISPRi was used over CRISPR/Cas9 knockout because genetic loss-of-function strategies are often complicated by transcriptional adaptation 71 , 72 . ( c ) Assessment of the activity of the mTORC1 and GCN2 pathways in low amino acid screen-like conditions for Arg, Lys, and His via Western blotting of downstream targets eIF2α (GCN2), and S6 kinase and 4E-BP1 (mTORC1). K562 cells grown in complete RPMI medium (Rich; the triangle depicts a centrifugation step) were rapidly pelleted and transferred to either Rich, low amino acid RPMI, or single amino acid dropout RPMI and samples were taken at specific time points after media exchange. The media were exchanged after 24 h (medium exchange denoted by asterisk). For all samples, cell lysates were immunoblotted with antibodies against vinculin (loading control), and phospho- and total antibodies against eIF2α, S6K, and 4E-BP1. ( d ) CRISPRi/a of transporters leads to specific changes in gene expression. Transporter expression levels in K562 CRISPRi/a cells with specific or non-targeting control (NTC) sgRNAs and grown in low Arg or low Leu RPMI was quantified by RT-qPCR relative to the housekeeping gene GAPDH. n = 3 technical replicates. Data are mean ± s.e.m. n.d., not detected. ( e ) low Arg and low Leu conditions induce a small transcriptional upregulation of most tested SLC7 transporters. Data are a subset of expression levels determined in Fig. 1b and ( d ). For each gene, expression levels of CRISPRi/a cell lines targeting that gene and NTC cell lines were plotted. Source numerical data and unprocessed blots are available in source data.
Extended Data Fig. 2 CRISPRi/a transporter screens in low amino acid conditions.
( a ) CRISPRi/a of transporters leads to changes in protein level at the plasma membrane. Changes in low Leu (L) and low Arg (R) conditions are similar to those observed in RPMI (rich). K562 CRISPRi/a cells were incubated with a cell-impermeable biotinylation reagent, and plasma membrane proteins were isolated by streptavidin affinity purification and analyzed by Western blotting. * denotes a non-specific band. ( b ) Tile plots displaying all significant transporter CRISPRi/a hits in low amino acid screens. Screen scores were calculated by multiplying phenotype scores by -log10( P value) for all genes and computed negative controls. P values were determined using a Mann-Whitney test. A stringent score cutoff was determined by the highest scoring negative control across all conditions for both datasets. All transporter genes that were significant in at least one condition tested were included in the tile plot. Transporter CRISPRi/a cell lines that have a growth defect in RPMI were prone to being pan-resistant in low amino acid conditions, as previously reported in other screens 25 , and were removed from the analysis (see Methods). Columns were ordered based on hierarchical clustering of scores. ( c ) CRISPRi/a transporter screens are highly reproducible as shown by the strong correlation between independent screen replicates. Black circles represent individual transporter genes and red circles indicate negative control genes. ( d ) The custom sgRNA library is highly homogenous, and the library preparation introduces no sample bias. gDNA was isolated from a pool of K562 CRISPRi transporter library cells post-antibiotic selection, and the abundance of each sgRNA present in the library was determined after PCR amplification and high-throughput sequencing (Methods). For this representative sample, >97% of sgRNAs were within 1 log2 of the mean, 12 sgRNAs had less than 1000 counts, and no sgRNA had 0 counts. Source numerical data and unprocessed blots are available in source data.
Extended Data Fig. 3 Measurement of amino acid import and consumption rates in K562 cells.
( a ) A cartoon illustrating the competition assay used to validate growth phenotypes of CRISPRi/a cell lines. A CRISPRi/a cell line expressing a test sgRNA is mixed at a 1:1 ratio with a cell line expressing a non-targeting control (NTC) sgRNA. Cell line ratios pre- and post-treatment are determined by qPCR on gDNA extracted from cultures using primers specific to the sgRNA. Phenotype scores are determined by normalizing enrichments by the difference in population doublings. ( b ) Confirmation of the specificity of primers targeting sgRNAs in qPCR assays. gDNA extracted from K562 CRISPRi/a cells was amplified by qPCR with primers targeting specific and NTC sgRNA barcodes. Data are the cycle threshold (Ct) value of a representative experiment. Asterisks represent non-specific product amplification. ( c ) SLC7A8 expression level in K562 SLC7A8 CRISPRi/a cells determined by RT-qPCR relative to the housekeeping gene GAPDH. n = 3 technical replicates. Data are mean ± s.e.m. ( d ) Cartoons illustrating amino acid import and consumption rate determination. The medium surrounding K562 cells attached to the surface of a Petri dish is rapidly exchanged to a similar medium where amino acids are heavy-isotope labelled. After extensive washing with PBS, intracellular metabolites are extracted and light and heavy amino acid levels are quantified by GC-MS. Import rates are determined from the slope of the increase in heavy amino acids over time. For consumption rate determination, the medium surrounding K562 cells is exchanged to fresh unlabelled medium and media samples are taken over time. Amino acid levels in samples are determined by GC-MS, and consumption rates are determined by the slope of the change in levels over time. ( e ) External standard curve used to calculate absolute levels of amino acids. Representative example of two independent experiments ( f ) Representative example of amino acid consumption from the medium of K562 cells growing in RPMI. Data (n = 7 biologically independent samples) were fit to a linear model and the shaded area represents 95% CI. ( g ) Numerical values of data displayed in Fig. 2c,d . Source numerical data are available in source data.
Extended Data Fig. 4 SLC7A5 CRISPRi specifically reduces transport of large neutral amino acids.
( a ) Comparison of amino acid import rates and inferred export rates in K562 cells growing in RPMI. Import rates are from Fig. 2c , and export rates were calculated by subtracting consumption rates from import rates. ( b , c ) Additional data related to Fig. 2f,i for amino acids that are not SLC7A5 substrates. ( d ) Comparison of amino acid import rates in low leucine RPMI and in RPMI for K562 SLC7A5 and non-targeting control (NTC) CRISPRi determined in Fig. 2f . ( e ) Comparison of intracellular amino acid levels for K562 SLC7A5 and NTC CRISPRi in low leucine RPMI and in RPMI determined in Fig. 2i . ( d , e ) Asterisks indicate SLC7A5 substrates. Source numerical data are available in source data.
Extended Data Fig. 5 Specific changes to amino acid levels and transport rates induced by CRISPRi/a of SLCs.
( a ) Changes in intracellular amino acid levels induced by CRISPRi/a of SLC7A1 and SLC7A7. ( b ) Same as (a) but for SLC38A3 CRISPRa and data from Fig. 3d . ( c ) LAT1–4 expression levels in K562 CRISPRi/a cell lines were determined by RT-qPCR relative to the housekeeping gene GAPDH. n = 3 technical replicates. Data are mean ± s.e.m. ( d ) Amino acid import rates into K562 cells correlate with levels of amino acid in the growth medium. Import rates for K562 CRISPRa SLC43A1 and non-targeting control (NTC) in RPMI and PAA–RPMI were from Fig. 3f . Data represent the ratio of import rates in PAA–RPMI to that in RPMI. Relative amino acid levels in the media were determined from their respective formulation. ( e ) The import of amino acids into K562 cells in low amino acid medium is selectively diminished for that specific low abundance amino acid. Data: mean ± s.e.m. of n = 2 (low Val), n = 3 (low Leu), n = 4 (RPMI) independent import rate determinations each calculated from the linear regression of n = 6 biologically independent samples. ( f ) SLC43A2 CRISPRa increases import of isoleucine and valine into K562 cells in RPMI. Rates were determined from a linear regression of n = 6 biologically independent samples. Data represent the slope ± SE normalized to NTC. Data for SLC43A1 CRISPRa is from Fig. 3f . ( g ) SLC43A2 CRISPRa induces a decrease in intracellular levels of large neutral amino acids in K562 cells grown in RPMI. ( h ) SLC43A1 CRISPRa leads to higher export of valine and similar export of leucine, isoleucine, and phenylalanine despite lower intracellular pools. Cells grown in RPMI containing heavy-isotope labelled amino acids were rapidly washed, then incubated in regular RPMI. Accumulation of heavy-labelled amino acids in RPMI was monitored over time by GC-MS. Representative example of 3 independent experiments. ( a , b , d , g ) n = 7 biologically independent samples. Data are mean ± s.e.m. Source numerical data are available in source data.
Extended Data Fig. 6 Specific changes to expression level of SLCs by CRISPRi/a.
( a ) RT-qPCR analysis shows strong and specific gene upregulation by CRISPRa. n = 3 technical replicates. Data are mean ± s.e.m. ( b ) SLC1A2 CRISPRa confers a growth advantage in low and no glutamine RPMI in the presence or absence of either glutamate or aspartate in the medium. Proliferation rates were extracted from competition assays with 4 biologically independent samples from two independent sgRNAs. P values were determined using two-tailed unpaired Student’s t-tests. ( c ) Expression level of SLC6A4 in cell lines used in this study and specific up- and down-regulation of SLC6A4 via CRISPRi/a. Levels of SLC6A4 and SLC7A11 were quantified by RT-qPCR relative to the housekeeping gene GAPDH in K562 CRISPRa, A375 CRISPRa, and Caco-2 CRISPRi cell lines with sgRNAs targeting SLC6A4, SLC7A11 or a non-targeting control (NTC). n = 3 technical replicates, except for A375 and Caco-2 NTC where n = 6. Data are mean ± s.e.m. ( d ) Violin plots displaying the distribution of lipid peroxidation levels in single cells (~20k) as determined by flow cytometry of cells incubated with BODIPY 581/591 C11 sensor after growth in the mentioned conditions. Red bars represent the average peroxidation level of the population and were used in Fig. 4g . ( e ) Low cystine induces expression of SLC7A11 but not of SLC6A4. K562 and Caco-2 cells were grown in either RPMI or low cystine RPMI for 3 days with daily media changes. Expression levels were quantified by RT-qPCR relative to GAPDH (n = 3 technical replicates. Data are mean ± s.e.m.). Source numerical data are available in source data.
Extended Data Fig. 7 Transporter essentiality across conditions.
( a ) CRISPRi transporter screens are highly reproducible as shown by the strong correlation of screen scores between independent replicates. Black circles represent individual transporter genes and red circles indicate computed negative control pseudogenes assembled from non-targeting control (NTC) sgRNAs. ( b ) Comparison of essential transporter genes identified in this study to previous screens in K562 cells grown in RPMI. The strong enrichment in expressed genes (98%) amongst the essential transporters in this study highlights the quality of the dataset. Essential transporters were determined by using a significance cutoff of q-value < 0.05 (ref. 37 ), and p < 0.05 and CS score negative 73 for the two CRISPRko screens (KO: knockout), and the first negative control gene for the genome-wide CRISPRi screen 34 (KD: knockdown). Genes were binned by expression level using publicly available data 37 . ( c ) Pairwise comparison of transporter growth scores determined in this study and in a genome-wide CRISPRi screen 34 in the same cell line, medium, and using the same gRNA library. ( d , e ) Transporter expression levels in K562 CRISPRi cell lines determined by RT-qPCR and relative to the housekeeping gene GAPDH (n = 3 technical replicates. Data are mean ± s.e.m.). ( f ) Growth scores of K562 MPC1 and MPC2 CRISPRi determined in screens in different media and the concentration of a selection of metabolites in those media. Screen scores are from Figs. 5c , 6c and metabolite levels are from published data 22 . ( g ) Addition of pyruvate to RPMI + FBS or RPMI + dialyzed FBS (dFBS) alleviates the growth defect induced by CRISPRi of MPC1 or MPC2, and addition of lactate to RPMI + dFBS worsens the growth defect. Assays were performed as in Fig. 5g and data were normalized to untreated samples. n = 6 biologically independent samples. Data are mean ± s.e.m. P values were determined using two-tailed unpaired Student’s t-tests. ( h ) Expression level of SFXN1 in K562 and A375 CRISPRi cell lines determined by RT-qPCR and relative to GAPDH (n = 3 technical replicates. Data are mean ± s.e.m.). Source numerical data are available in source data.
Extended Data Fig. 8 Optimization of subcutaneous CRISPRi/a transporter screens.
( a ) Determination of the engraftment frequency of K562 cells injected subcutaneously into immunodeficient mice. K562 CRISPRi library cells expressing GFP were mixed into pools of K562 CRISPRi library cells at a ratio ranging from 1:1000–1:100,000. 10, 1 or 0.1 million cells of these preparations were injected into the flanks of mice. Tumors formed over 19–34 days, were harvested and dissociated into single cell suspensions. The presence (or absence) of GFP + cells in homogenized tumor samples was determined by flow cytometry and was used to determine the engraftment frequency and conditions required to preserve library complexity during in vivo transporter screens. ( b ) Plot displaying the number of counts for all non-targeting control (NTC) sgRNAs determined either from the sequencing of tumor samples from (a), from the initial pool of library cells, or from library cells grown in RPMI. ( c ) Expression level of SLC2A1 in K562 and A375 CRISPRa cell lines determined by RT-qPCR and relative to the housekeeping gene GAPDH. n = 3 technical replicates. Data are mean ± s.e.m. ( d ) Overexpression of SLC2A1 via CRISPRa leads to increased levels at the plasma membrane in K562 and A375 cells. Cells were incubated with a cell-impermeable biotinylation reagent, and plasma membrane proteins were isolated by streptavidin affinity purification and analyzed by Western blotting. ( e ) Example of gating strategy used for flow cytometry sorting of K562 cells transduced with lentivirus expressing dCas9-BFP-KRAB for K562 CRISPRi/a cell line generation. Source numerical data and unprocessed blots are available in source data.
Supplementary information
Reporting summary, peer review file, supplementary table 1.
Transporter CRISPRi and CRISPRa sgRNA libraries.
Supplementary Table 2
Growth media used in this study.
Supplementary Table 3
Amino acid levels and population doublings in CRISPRi/a screens.
Supplementary Table 4
Oligonucleotides used in this study.
Supplementary Table 5
CRISPRi screen scores.
Supplementary Table 6
CRISPRa screen scores.
Source Data Fig. 1
Numerical source data.
Source Data Fig. 2
Source data fig. 3, source data fig. 4, source data fig. 5, source data fig. 6, source data extended data fig. 1, source data extended data fig. 2, source data extended data fig. 3, source data extended data fig. 4, source data extended data fig. 5, source data extended data fig. 6, source data extended data fig. 7, source data extended data fig. 8, source data.
Unprocessed western blots. One file for all blots.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ .
Reprints and permissions
About this article
Cite this article.
Chidley, C., Darnell, A.M., Gaudio, B.L. et al. A CRISPRi/a screening platform to study cellular nutrient transport in diverse microenvironments. Nat Cell Biol 26 , 825–838 (2024). https://doi.org/10.1038/s41556-024-01402-1
Download citation
Received : 23 May 2023
Accepted : 07 March 2024
Published : 11 April 2024
Issue Date : May 2024
DOI : https://doi.org/10.1038/s41556-024-01402-1
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Harnessing the evolving crispr/cas9 for precision oncology.
- Shuiquan Li
Journal of Translational Medicine (2024)
Ferroptosis: principles and significance in health and disease
- Fangquan Chen
Journal of Hematology & Oncology (2024)
Exploring the potential of asparagine restriction in solid cancer treatment: recent discoveries, therapeutic implications, and challenges
- Marina Gabriel Fontes
- Carolina Silva
- Gisele Monteiro
Medical Oncology (2024)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Cancer newsletter — what matters in cancer research, free to your inbox weekly.


Want to create or adapt books like this? Learn more about how Pressbooks supports open publishing practices.
Chapter 8. Membrane Transport

Chapter Outline
- 8.1 Membrane Components and Structure
- 8.2 Passive Transport
- 8.3 Active Transport
- 8.4 Bulk Transport
Introduction
The plasma membrane, which is also called the cell membrane, has many functions, but the most basic one is to define the borders of the cell and keep the cell functional. The plasma membrane is selectively permeable. This means that the membrane allows some materials to freely enter or leave the cell, while other materials cannot move freely, but require the use of a specialized structure, and occasionally, even energy investment for crossing.
8.1 | Membrane Components and Structure
Learning Objectives
By the end of this section, you will be able to:
- Understand the fluid mosaic model of cell membranes.
- Describe the functions of phospholipids, proteins, and carbohydrates in membranes.
- Discuss membrane fluidity.
A cell’s plasma membrane defines the cell, outlines its borders, and determines the nature of its interaction with its environment (see Table 8.1 for a summary). Cells exclude some substances, take in others, and excrete still others, all in controlled quantities. The plasma membrane must be very flexible to allow certain cells, such as red blood cells and white blood cells, to change shape as they pass through narrow capillaries. These are the more obvious functions of a plasma membrane. In addition, the surface of the plasma membrane carries markers that allow cells to recognize one another, which is vital for tissue and organ formation during early development, and which later plays a role in the “self” versus “non-self” distinction of the immune response.
Among the most sophisticated functions of the plasma membrane is the ability to transmit signals by means of complex, integral proteins known as receptors. These proteins act both as receivers of extracellular inputs and as activators of intracellular processes. These membrane receptors provide extracellular attachment sites for effectors like hormones and growth factors, and they activate intracellular response cascades when their effectors are bound. Occasionally, receptors are hijacked by viruses (HIV, human immunodeficiency virus, is one example) that use them to gain entry into cells, and at times, the genes encoding receptors become mutated, causing the process of signal transduction to malfunction with disastrous consequences.
8.1.1 Fluid Mosaic Model
The existence of the plasma membrane was identified in the 1890s, and its chemical components were identified in 1915. The principal components identified at that time were lipids and proteins. The first widely accepted model of the plasma membrane’s structure was proposed in 1935 by Hugh Davson and James Danielli; it was based on the “railroad track” appearance of the plasma membrane in early electron micrographs. They theorized that the structure of the plasma membrane resembles a sandwich, with protein being analogous to the bread, and lipids being analogous to the filling. In the 1950s, advances in microscopy, notably transmission electron microscopy (TEM), allowed researchers to see that the core of the plasma membrane consisted of a double, rather than a single, layer. A new model that better explains both the microscopic observations and the function of that plasma membrane was proposed by S.J. Singer and Garth L. Nicolson in 1972.
The explanation proposed by Singer and Nicolson is called the fluid mosaic model . The model has evolved somewhat over time, but it still best accounts for the structure and functions of the plasma membrane as we now understand them. The fluid mosaic model describes the structure of the plasma membrane as a mosaic of components—including phospholipids, cholesterol, proteins, and carbohydrates—that gives the membrane a fluid character. Plasma membranes range from 5 to 10 nm in thickness. For comparison, human red blood cells, visible via light microscopy, are approximately 8 µm wide, or approximately 1,000 times wider than a plasma membrane. The membrane does look a bit like a sandwich ( Figure 8.2 ).

The principal components of a plasma membrane are lipids, proteins, and carbohydrates. The lipids include phospholipids and cholesterol Proteins either float in the bilayer or are attached to one side or the other of it. Carbohydrate chains are attached to the proteins and lipids on the outside surface of the membrane. The proportions of proteins, lipids, and carbohydrates in the plasma membrane vary with cell type, but for a typical human cell, protein accounts for about 50 percent of the composition by mass, lipids account for about 40 percent of the composition by mass, with the remaining 10 percent of the composition by mass being carbohydrates.
Phospholipids
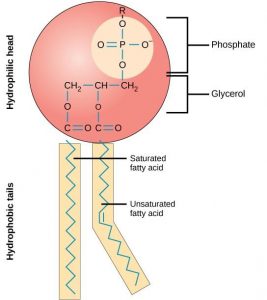
The main fabric of the membrane is composed of amphiphilic phospholipid molecules. Recall from chapter 4 that a phospholipid is a molecule consisting of glycerol, two fatty acids, and a phosphate-linked head group ( Figure 8.3 ) . The hydrophilic “head” of these molecules are in contact with the aqueous fluid both inside and outside the cell. The hydrophobic “tails” face each other in the inside of the bilayer. Therefore, phospholipids form an excellent two-layer cell membrane that separates fluid within the cell from the fluid outside of the cell ( Figure 8.2 ).
The amphipathic nature of phospholipids is vital to the structure of a plasma membrane because, in water, phospholipids automatically become arranged with their hydrophobic tails facing each other and their hydrophilic heads facing out. In this way, they form a lipid bilayer—a barrier composed of a double layer of phospholipids that separates the water and other materials on one side of the barrier from the water and other materials on the other side ( Figure 8 .4 top ). In fact, phospholipids heated in an aqueous solution tend to spontaneously form small spheres or droplets called micelles, with their hydrophilic heads forming the exterior and their hydrophobic tails on the inside ( Figure 8 .4 bottom ).

Proteins make up the second major component of plasma membranes. Integral proteins are, as their name suggests, integrated completely into the membrane structure, and their hydrophobic membrane-spanning regions interact with the hydrophobic region of the the phospholipid bilayer ( Figure 8.2 ). Single-pass integral membrane proteins usually have a hydrophobic transmembrane segment that consists of 20–25 amino acids. Some span only part of the membrane— associating with a single layer—while others stretch from one side of the membrane to the other, and are exposed on either side. Since they cross the membrane, these are often called transmembrane proteins .
Some complex integral proteins are composed of up to 12 segments, which are extensively folded and embedded in the membrane ( Figure 8.5 ). This type of protein has a hydrophilic region or regions, and several hydrophobic regions. This arrangement of regions of the protein tends to orient the protein alongside the phospholipids, with the hydrophobic region of the protein adjacent to the tails of the phospholipids and the hydrophilic region or regions of the protein protruding from the membrane and in contact with the cytosol or extracellular fluid.

Peripheral proteins are found on the exterior and interior surfaces of membranes, attached either to integral proteins or to phospholipids. Peripheral proteins, along with integral proteins, may serve as enzymes, as structural attachments for the fibers of the cytoskeleton, or as part of the cell’s recognition sites. These are sometimes referred to as “cell-specific” proteins. The body recognizes its own proteins and attacks foreign proteins associated with invasive pathogens.
Carbohydrates
Carbohydrates are the third major component of plasma membranes. They are always found on the exterior surface of cells and are bound either to proteins (forming glycoproteins ) or to lipids (forming glycolipids ) ( Figure 8.2 ). These carbohydrate chains may consist of 2–60 monosaccharide units and can be either straight or branched. Along with peripheral proteins, carbohydrates form specialized sites on the cell surface that allow cells to recognize each other. These sites have unique patterns that allow the cell to be recognized, much the way that the facial features unique to each person allow him or her to be recognized. This recognition function is very important to cells, as it allows the immune system to differentiate between body cells (called “self”) and foreign cells or tissues (called “non-self”). Similar types of glycoproteins and glycolipids are found on the surfaces of viruses and may change frequently, preventing immune cells from recognizing and attacking them.
These carbohydrates on the exterior surface of the cell—the carbohydrate components of both glycoproteins and glycolipids—are collectively referred to as the glycocalyx (meaning “sugar coating”). The glycocalyx is highly hydrophilic and attracts large amounts of water to the surface of the cell. This aids in the interaction of the cell with its watery environment and in the cell’s ability to obtain substances dissolved in the water. As discussed above, the glycocalyx is also important for cell identification, self/non-self determination, and embryonic development, and is used in cell-cell attachments to form tissues.
How Viruses Infect Specific Organs
Glycoprotein and glycolipid patterns on the surfaces of cells give many viruses an opportunity for infection. HIV and hepatitis viruses infect only specific organs or cells in the human body. HIV is able to penetrate the plasma membranes of a subtype of lymphocytes called T-helper cells, as well as some monocytes and central nervous system cells. The hepatitis virus attacks liver cells.
These viruses are able to invade these cells, because the cells have binding sites on their surfaces that are specific to and compatible with certain viruses ( Figure 8 .6 ). Other recognition sites on the virus’s surface interact with the human immune system, prompting the body to produce antibodies. Antibodies are made in response to the antigens or proteins associated with invasive pathogens, or in response to foreign cells, such as might occur with an organ transplant. These same sites serve as places for antibodies to attach and either destroy or inhibit the activity of the virus. Unfortunately, these recognition sites on HIV change at a rapid rate because of mutations, making the production of an effective vaccine against the virus very difficult, as the virus evolves and adapts. A person infected with HIV will quickly develop different populations, or variants, of the virus that are distinguished by differences in these recognition sites. This rapid change of surface markers decreases the effectiveness of the person’s immune system in attacking the virus, because the antibodies will not recognize the new variations of the surface patterns. In the case of HIV, the problem is compounded by the fact that the virus specifically infects and destroys cells involved in the immune response, further incapacitating the host.
8.1.2 Membrane Fluidity
The mosaic characteristic of the membrane, described in the fluid mosaic model, helps to illustrate its nature. The integral proteins and lipids exist in the membrane as separate but loosely attached molecules. These resemble the separate, multicolored tiles of a mosaic picture, and they float, moving somewhat with respect to one another. The membrane is not like a balloon, however, that can expand and contract; rather, it is fairly rigid and can burst if penetrated or if a cell takes in too much water. However, because of its mosaic nature, a very fine needle can easily penetrate a plasma membrane without causing it to burst, and the membrane will flow and self-seal when the needle is extracted.
The mosaic characteristics of the membrane explain some but not all of its fluidity. There are two other factors that help maintain this fluid characteristic. One factor is the nature of the phospholipids themselves. In their saturated form, the fatty acids in phospholipid tails are saturated with bound hydrogen atoms. There are no double bonds between adjacent carbon atoms. This results in tails that are relatively straight. In contrast, unsaturated fatty acids do not contain a maximal number of hydrogen atoms, but they do contain some double bonds between adjacent carbon atoms; a double bond results in a bend in the string of carbons of approximately 30 degrees ( Figure 8.3 ).
Thus, if saturated fatty acids, with their straight tails, are compressed by decreasing temperatures, they press in on each other, making a dense and fairly rigid membrane. If unsaturated fatty acids are compressed, the “kinks” in their tails elbow adjacent phospholipid molecules away, maintaining some space between the phospholipid molecules. This “elbow room” helps to maintain fluidity in the membrane at temperatures at which membranes with saturated fatty acid tails in their phospholipids would “freeze” or solidify. The relative fluidity of the membrane is particularly important in a cold environment. A cold environment tends to compress membranes composed largely of saturated fatty acids, making them less fluid and more susceptible to rupturing. Many organisms (fish are one example) are capable of adapting to cold environments by changing the proportion of unsaturated fatty acids in their membranes in response to the lowering of the temperature.
Animals have an additional membrane constituent that assists in maintaining fluidity. Cholesterol, which lies alongside the phospholipids in the membrane, tends to dampen the effects of temperature on the membrane. Thus, this lipid functions as a buffer, preventing lower temperatures from inhibiting fluidity and preventing increased temperatures from increasing fluidity too much. Thus, cholesterol extends, in both directions, the range of temperature in which the membrane is appropriately fluid and consequently functional. Cholesterol also serves other functions, such as organizing clusters of transmembrane proteins into lipid rafts.
Table 8.1 The components and functions of the plasma membrane.
|
|
|
| Phospholipid | Main fabric of the membrane |
| Cholesterol | Attached between phospholipids and between the two phospholipid layers |
| Integral proteins (for example, integrins) | Embedded within the phospholipid layer(s). May or may not penetrate through both layers |
| Peripheral proteins | On the inner or outer surface of the phospholipid bilayer; not embedded within the phospholipids |
| Carbohydrates (components of glycoproteins and glycolipids) | Generally attached to proteins on the outside membrane layer |
Immunologist
The variations in peripheral proteins and carbohydrates that affect a cell’s recognition sites are of prime interest in immunology. These changes are taken into consideration in vaccine development. Many infectious diseases, such as smallpox, polio, diphtheria, and tetanus, were conquered by the use of vaccines.
dImmunologists are the physicians and scientists who research and develop vaccines, as well as treat and study allergies or other immune problems. Some immunologists study and treat autoimmune problems (diseases in which a person’s immune system attacks his or her own cells or tissues, such as lupus) and immunodeficiencies, whether acquired (such as acquired immunodeficiency syndrome, or AIDS) or hereditary (such as severe combined immunodeficiency, or SCID). Immunologists are called in to help treat organ transplantation patients, who must have their immune systems suppressed so that their bodies will not reject a transplanted organ. Some immunologists work to understand natural immunity and the effects of a person’s environment on it. Others work on questions about how the immune system affects diseases such as cancer. In the past, the importance of having a healthy immune system in preventing cancer was not at all understood.
To work as an immunologist, a PhD or MD is required. In addition, immunologists undertake at least 2–3 years of training in an accredited program and must pass an examination given by the American Board of Allergy and Immunology. Immunologists must possess knowledge of the functions of the human body as they relate to issues beyond immunization, and knowledge of pharmacology and medical technology, such as medications, therapies, test materials, and surgical procedures.
8.2 | Passive Transport
- Explain why and how passive transport occurs.
- Understand the processes of osmosis and diffusion.
- Define tonicity and describe its relevance to passive transport.
Plasma membranes must allow certain substances to enter and leave a cell, and prevent some harmful materials from entering and some essential materials from leaving. In other words, plasma membranes are selectively permeable —they allow some substances to pass through, but not others. If they were to lose this selectivity, the cell would no longer be able to sustain itself, and it would be destroyed. Some cells require larger amounts of specific substances than do other cells; they must have a way of obtaining these materials from extracellular fluids. This may happen passively, as certain materials move back and forth, or the cell may have special mechanisms that facilitate transport. Some materials are so important to a cell that it spends some of its energy, hydrolyzing adenosine triphosphate (ATP), to obtain these materials. All cells spend the majority of their energy to maintain an imbalance of sodium and potassium ions between the interior and exterior of the cell.
The most direct forms of membrane transport are passive. Passive transport is a naturally occurring phenomenon and does not require the cell to exert any of its energy to accomplish the movement. In passive transport, substances move from an area of higher concentration to an area of lower concentration. A physical space in which there is a range of concentrations of a single substance is said to have a concentration gradient .
8.2.1 Selective Permeability
Plasma membranes are asymmetric: the interior of the membrane is not identical to the exterior of the membrane. In fact, there is a considerable difference between the array of phospholipids and proteins between the two leaflets that form a membrane. On the interior of the membrane, some proteins serve to anchor the membrane to fibers of the cytoskeleton. There are peripheral proteins on the exterior of the membrane that bind elements of the extracellular matrix. Carbohydrates,
attached to lipids or proteins, are also found on the exterior surface of the plasma membrane. These carbohydrate complexes help the cell bind substances that the cell needs in the extracellular fluid. This adds considerably to the selective nature of plasma membranes ( Figure 8.7 ).

Recall that plasma membranes are amphipathic: They have hydrophilic and hydrophobic regions. This characteristic helps the movement of some materials through the membrane and hinders the movement of others. Lipid-soluble material with a low molecular weight can easily slip through the hydrophobic lipid core of the membrane. Substances such as the fat- soluble vitamins A, D, E, and K readily pass through the plasma membranes in the digestive tract and other tissues. Fat-soluble drugs and hormones also gain easy entry into cells and are readily transported into the body’s tissues and organs. Molecules of oxygen and carbon dioxide have no charge and so pass through membranes by simple diffusion.
Polar substances present problems for the membrane. While some polar molecules connect easily with the outside of a cell, they cannot readily pass through the lipid core of the plasma membrane. Additionally, while small ions could easily slip through the spaces in the mosaic of the membrane, their charge prevents them from doing so. Ions such as sodium, potassium, calcium, and chloride must have special means of penetrating plasma membranes. Larger polar molecules, such as simple sugars and amino acids also need help with transport across plasma membranes.
8.2.2 Diffusion
Diffusion is a passive process of transport. A single substance tends to move from an area of high concentration to an area of low concentration until the concentration is equal across a space. You are familiar with diffusion of substances through the air. For example, think about someone opening a bottle of ammonia in a room filled with people. The ammonia gas is at its highest concentration in the bottle; its lowest concentration is at the edges of the room. The ammonia vapor will diffuse, or spread away, from the bottle, and gradually, more and more people will smell the ammonia as it spreads. Materials move within the cell’s cytosol by diffusion, and certain materials move through the plasma membrane by diffusion ( Figure 8.8 ). Diffusion expends no energy. On the contrary, concentration gradients are a form of potential energy, dissipated as the gradient is eliminated.
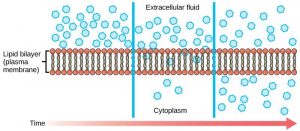
Each separate substance in a medium, such as the extracellular fluid, has its own concentration gradient, independent of the concentration gradients of other materials. In addition, each substance will diffuse according to that gradient. Within a system, there will be different rates of diffusion of the different substances in the medium.
Factors That Affect Diffusion
Molecules move constantly in a random manner, at a rate that depends on their mass, their environment, and the amount of thermal energy they possess, which in turn is a function of temperature. This movement accounts for the diffusion of molecules through whatever medium in which they are localized. A substance will tend to move into any space available to it until it is evenly distributed throughout it. After a substance has diffused completely through a space, removing its concentration gradient, molecules will still move around in the space, but there will be no net movement of the number of molecules from one area to another. This lack of a concentration gradient in which there is no net movement of a substance is known as dynamicequilibrium . While diffusion will go forward in the presence of a concentration gradient of a substance, several factors affect the rate of diffusion.
“Steepness” of the concentration gradient: The greater the difference in concentration, the more rapid the diffusion. The closer the distribution of the material gets to equilibrium, the slower the rate of diffusion becomes.
Mass of the molecules diffusing: Heavier molecules move more slowly; therefore, they diffuse more slowly.
Temperature: Higher temperatures increase the energy and therefore the movement of the molecules, increasing the rate of diffusion.
Solvent density: As the density of a solvent increases, the rate of diffusion decreases. The molecules slow down because they have a more difficult time getting through the denser medium. If the medium is less dense, diffusion increases. Because cells primarily use diffusion to move materials within the cytoplasm, any increase in the cytoplasm’s density will inhibit the movement of the materials. An example of this is a person experiencing dehydration. As the body’s cells lose water, the rate of diffusion decreases in the cytoplasm, and the cells’ functions deteriorate. Neurons tend to be very sensitive to this effect. Dehydration frequently leads to unconsciousness and possibly coma because of the decrease in diffusion rate within the cells.
8.2.3 Facilitated diffusion
In facilitated diffusion , materials diffuse across the plasma membrane with the help of membrane proteins. A concentration gradient exists that would allow these materials to diffuse into the cell without expending cellular energy. However, these materials are ions or polar molecules that are repelled by the hydrophobic parts of the cell membrane. Facilitated diffusion proteins shield these materials from the repulsive force of the membrane, allowing them to diffuse into the cell. These proteins are called transportproteins and can be channels or carrier proteins.
Channelproteins are transmembrane proteins that fold in such as way as to form a channel or pore through the membrane. Each channel is specific for one particular substance. Channel proteins have hydrophilic domains exposed to the intracellular and extracellular fluids. In addition, they have a hydrophilic channel through their core that provides a hydrated opening through the membrane layers ( Figure8.9 ). Passage through the channel allows polar compounds to avoid the nonpolar central layer of the plasma membrane that would otherwise slow or prevent their entry into the cell. Aquaporins are channel proteins that allow water to pass through the membrane at a very high rate.
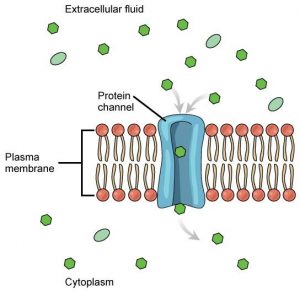
Some channel proteins are always open but many are “gated,” meaning that they can be opened and closed. If a channel is ligand-gated, the attachment of a particular molecule to the channel protein may cause it to open. Other channels are voltage-gated, requiring a change in voltage across the membrane to open them. Cells involved in the transmission of electrical impulses, such as nerve and muscle cells, have voltage-gated ion channels in their membranes.
Carrier Proteins
Another type of transmembrane transporter protein is a carrierprotein . Like channels, carrier proteins are usually specific for particular molecules. A carrier proteins binds a substance and, in doing so, triggers a change of its own shape, moving the bound molecule across the membrane ( Figure8.10 ). Carrier proteins are used to transport molecules that are too large to pass through channels, such as amino acids and glucose.

There are a finite number of each type of carrier proteins in any membrane. This can cause problems in transporting enough of the material for the cell to function properly. When all of the proteins are bound to their ligands, they are saturated and the rate of transport is at its maximum. Increasing the concentration gradient at this point will not result in an increased rate of transport.
An example of this process occurs in the kidney. Glucose, water, salts, ions, and amino acids needed by the body are filtered out of the blood in one part of the kidney. This filtrate, which includes glucose, is then reabsorbed in another part of the kidney. Because there are only a finite number of carrier proteins for glucose, if more glucose is present than the proteins can handle, the excess is not transported and it is excreted from the body in the urine. In a diabetic individual, this is described as “spilling glucose into the urine.”
A different group of carrier proteins called glucose transport proteins, or GLUTs, are involved in transporting glucose and other hexose sugars into cells within the body. The hormone insulin, increases the number of GLUTs on cells, causing them to take glucose from the blood when its levels are high. It is this process that is compromised in diabetic individuals.
Channel proteins transport much more quickly than do carrier proteins. Channel proteins facilitate diffusion at a rate of tens of millions of molecules/second, whereas carrier proteins work at a rate of a thousand to a million molecules/second.
8.2.4 Osmosis
Osmosis is the diffusion of water across a semipermeable membrane. Since it is diffusion, it depends on the concentration gradient, or the amount of water on each side of the membrane. The amount of water in a solute is inversely proportional to the concentration of solutes. In other words, the higher the concentration of water, the lower the concentration of solutes, and vice versa. Water can move readily across most membranes, due in part to the presence of aquaporins; however, the membrane limits the diffusion of solutes in the water.
Mechanism of Osmosis
Osmosis is a special case of diffusion. Water, like other substances, moves from an area of high concentration to one of low concentration. An obvious question is what makes water move at all? Imagine a beaker with a semipermeable membrane separating the two sides or halves ( Figure8.11 ). On both sides of the membrane the water level is the same, but there are different concentrations of a dissolved substance, or solute , that cannot cross the membrane (otherwise the concentrations on each side would be balanced by the solute crossing the membrane). If the volume of the solution on both sides of the membrane is the same, but the concentrations of solute are different, then there are different amounts of water, the solvent, on either side of the membrane.
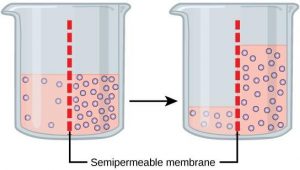
To illustrate this, imagine two full glasses of water. One has a single teaspoon of sugar in it, whereas the second one contains one-quarter cup of sugar. If the total volume of the solutions in both cups is the same, which cup contains more water? Because the large amount of sugar in the second cup takes up much more space than the teaspoon of sugar in the first cup, the first cup has more water in it.
Returning to the beaker example, recall that it has a mixture of solutes on either side of the membrane. A principle of diffusion is that the molecules move around and will spread evenly throughout the medium if they can. However, only the material capable of getting through the membrane will diffuse through it. In this example, the solute cannot diffuse through the membrane, but the water can. Water has a concentration gradient in this system. Thus, water will diffuse down its concentration gradient, crossing the membrane to the side where it is less concentrated. This diffusion of water through the membrane—osmosis—will continue until the concentration gradient of water goes to zero or until the hydrostatic pressure of the water balances the osmotic pressure. Osmosis proceeds constantly in living systems.
8.2.5 Tonicity
Tonicity describes how an extracellular solution can change the volume of a cell by affecting osmosis. A solution’s tonicity often directly correlates with the osmolarity of the solution. Osmolarity describes the total solute concentration of the solution. A solution with low osmolarity has a greater number of water molecules relative to the number of solute particles; a solution with high osmolarity has fewer water molecules with respect to solute particles. In a situation in which solutions of two different osmolarities are separated by a membrane permeable to water, though not to the solute, water will move from the side of the membrane with lower osmolarity (and more water) to the side with higher osmolarity (and less water). This effect makes sense if you remember that the solute cannot move across the membrane, and thus the only component in the system that can move—the water—moves along its own concentration gradient.
Three terms—hypotonic, isotonic, and hypertonic—are used to relate the osmolarity of a cell to the osmolarity of the extracellular fluid. In living systems, the point of reference is always the cytoplasm, so the prefix hypo- (“lower”) means that the extracellular fluid has a lower concentration of solutes, or a lower osmolarity, than the cell cytoplasm. Blood cells and plant cells in hypertonic, isotonic, and hypotonic solutions take on characteristic appearances ( Figure 8.12 ).
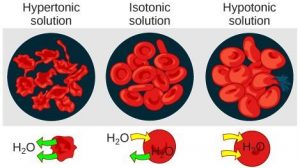
Hypotonic Solutions
In a hypotonic situation, the extracellular fluid has lower osmolarity than the fluid inside the cell. The extracellular fluid has a higher concentration of water than does the cell and water will move down its concentration gradient and enter the cell.
Hypertonic Solutions
In a hypertonic solution (hyper- = “more”), the extracellular fluid has a higher osmolarity than the cell’s cytoplasm. The fluid contains less water than the cell does, so water will leave the cell.
Isotonic Solutions
In an isotonic solution, the extracellular fluid has the same osmolarity as the cell. There is no net movement of water into or out of the cell (although water will still move in and out).
Concept Check
A doctor injects a patient with what the doctor thinks is an isotonic saline solution. The patient dies, and an autopsy reveals that many red blood cells have been destroyed. Do you think the solution the doctor injected was really isotonic?
8.2.6 Tonicity in Living Systems
A red blood cell will burst, or lyse, when it swells beyond the plasma membrane’s capability to expand. In contrast, when excessive amounts of water leave a red blood cell, the cell shrinks, or crenates. Crenation has the effect of concentrating the solutes left in the cell, making the cytosol denser and interfering with diffusion within the cell. The cell’s ability to function will be compromised and it may die. ( Figure 8.12 ).
Living things have ways of controlling the effects of osmosis—a mechanism called osmoregulation. Some organisms, such as plants, fungi, bacteria, and some protists, have cell walls that surround the plasma membrane and prevent cells from lysing. In fact, the cytoplasm in plants is always slightly hypertonic to the cellular environment, and water will always enter a cell if water is available. This inflow of water produces turgor pressure, which stiffens the cell walls of the plant ( Figure 8.13 ). In nonwoody plants, turgor pressure supports the plant. If the plant is not watered, the extracellular fluid will become hypertonic, causing water to leave the cell. In this condition, the cell membrane detaches from the cell wall and constricts the cytoplasm. This process, called plasmolysis , causes plants to lose turgor pressure ( Figure 8.14 ).

Tonicity is a concern for all living things. For example, paramecia and amoebas, which are protists that lack cell walls, have contractile vacuoles. This vesicle collects excess water from the cell and pumps it out, keeping the cell from lysing as it takes on water from its environment ( Figure 8.15 ).
Many marine invertebrates have internal salt levels matched to their environments, making them isotonic with the water in which they live. Fish, however, must spend approximately five percent of their metabolic energy maintaining osmotic homeostasis. Freshwater fish live in an environment that is hypotonic to their cells. These fish actively take in salt through their gills and excrete diluted urine to rid themselves of excess water. Saltwater fish live in the reverse environment, which is hypertonic to their cells, and they secrete salt through their gills and excrete highly concentrated urine.
In vertebrates, the kidneys regulate the amount of water in the body. Osmoreceptors are specialized cells in the brain that monitor the concentration of solutes in the blood. If the levels of solutes increase beyond a certain range, a hormone is released that retards water loss through the kidney and dilutes the blood to safer levels. Animals also have high concentrations of albumin, which is produced by the liver, in their blood. This protein is too large to pass easily through plasma membranes and is a major factor in controlling the osmotic pressures applied to tissues.
8.3 | Active Transport
By the end of this section, you will be able to:Understand how electrochemical gradients affect ionsDistinguish between primary active transport and secondary active transport
Active transport mechanisms require the use of the cell’s energy, usually in the form of adenosine triphosphate (ATP). If a substance must move into the cell against its concentration gradient—that is, if the concentration of the substance inside the cell is greater than its concentration in the extracellular fluid (and vice versa)—the cell must use energy to move the substance. Some active transport mechanisms move small-molecular weight materials, such as ions, through the membrane. Other mechanisms transport much larger molecules.
8.3.1 Electrochemical Gradient
We have discussed simple concentration gradients—different concentrations of a substance across a space or a membrane—but in living systems, gradients are more complex. Because ions move into and out of cells and because cells contain proteins that do not move across the membrane and are mostly negatively charged, there is also an electrical gradient, a difference of charge, across the plasma membrane.
The interior of living cells is electrically negative with respect to the extracellular fluid surrounding them. At the same time, cells have a lower concentration of (Na+) than does the extracellular fluid. Therefore, both the concentration
gradient and the electrical gradient tend to drive Na+ into the cell. Conversely, cells have a higher concentration of K+ than the extracellular fluid does. Therefore, the concentration gradient tends to drive K+ out of the cell, while the electrical gradient tends to drive it inside the cell. The combined gradient of concentration and electrical charge that affects an ion is called its electrochemical gradient ( Figure 8.16 ).
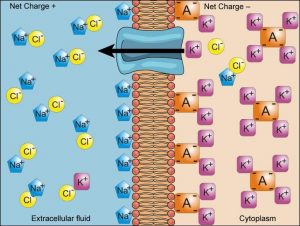
Injection of a potassium solution into a person’s blood is lethal; this is used in capital punishment and euthanasia. Why do you think a potassium solution injection is lethal?
Moving Against a Gradient
To move substances against a concentration or electrochemical gradient, the cell must use energy, usually in the form of ATP. Active transport proteins, called pumps , work against electrochemical gradients. Small substances constantly pass through plasma membranes. Active transport maintains concentrations of ions and other substances needed by living cells in the face of these passive movements. Much of a cell’s supply of metabolic energy may be spent maintaining these processes.
Proteins for Active Transport
The specific proteins that facilitate active transport are called transporters . There are three types of transporters ( Figure 8.17 ). A uniporter carries one specific ion or molecule. A symporter carries two different ions or molecules, both in the same direction. An antiporter carries two different ions or molecules in different directions. All of these transporters can transport small, uncharged organic molecules such as glucose.
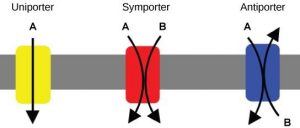
Two mechanisms exist for the transport of small-molecular weight material and small molecules. Primary active transport is directly dependent on ATP. Secondary active transport does not directly require ATP, because it uses electrochemical gradients established by primary active transport for fuel. Primary active transport must occur first to in order to allow secondary active transport to occur. Although it does not use ATP, secondary active transport is still considered active because it requires energy.
8.3.2 Primary Active Transport
One of the most important pumps in animals cells is the sodium-potassium pump (Na+-K+ ATPase), which maintains the electrochemical gradient and the correct concentrations of Na+ and K+ in living cells. The sodium-potassium pump moves two K+ into the cell while moving three Na+ out of the cell ( Figure 8.18 ).

The sodium-potassium pump works in the following six steps:
- Three sodium ions bind to the protein.
- ATP is hydrolyzed by the protein carrier and a low-energy phosphate group attaches to it.
- The carrier changes shape and opens towards the exterior of the membrane. The three sodium ions are released.
- Two potassium ions attach to the protein, causing the low-energy phosphate group to detach.
- The carrier protein changes shape so that is open towards the interior of the cell.
- The two potassium ions are released into the cytoplasm and the process begins again.
Several things have happened as a result of this process. First, there are now more sodium ions outside of the cell than inside and more potassium ions inside than out. Second, since three sodium ions moved out for each two potassium ions that moved in, the interior is slightly more negative relative to the exterior. This difference in charge is important in creating the conditions necessary for secondary active transport. The sodium-potassium pump is, therefore, an electrogenic pump (a pump that creates a charge imbalance), creating an electrical imbalance across the membrane and contributing to the membrane potential.
The sodium-potassium pump (Na+/K+ pump) is one example of energy coupling. Each cycle of the Na+/K+ pump moves three sodium out of the cell and brings two potassium into the cell. For each cycle, one ATP is hydrolyzed and its free phosphate group is transferred to the pump protein. This process of a phosphate group binding to a molecule is called phosphorylation . Phosphorylation of the pump protein causes it to change shape, moving ions across the membrane. ATP performs cellular work using this basic form of energy coupling through phosphorylation. Here, the exergonic (energy-releasing) process of ATP breakdown “pays for” the endergonic (energy-requiring) process of moving ions against their concentration gradients.
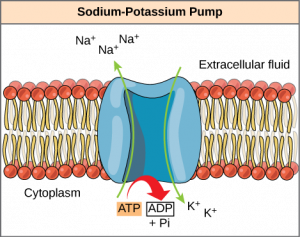
8.3.3 Secondary Active Transport (Co-transport)
Secondary active transport moves a solute against its concentration gradient, an endergonic process, by moving another solute down its concentration gradient, an exergonic process. For instance, as sodium ion concentrations build outside of the plasma membrane because of the action of the sodium-potassium pump, an electrochemical gradient is created. If a channel protein exists and is open, the sodium ions will be pulled through the membrane, down their concentration gradient. This exergonic movement is used to transport other substances that can attach themselves to the transport protein through the membrane ( Figure 8.20 ). Many amino acids, as well as glucose, enter a cell this way.
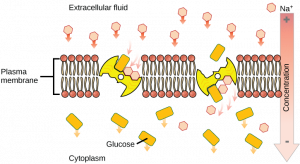
8.4 | Bulk Transport
- Describe endocytosis, including phagocytosis, pinocytosis, and receptor-mediated endocytosis.
- Understand the process of exocytosis.
In addition to moving small ions and molecules through the membrane, cells also need to remove and take in larger molecules and particles (see Table 8.2 for examples). Some cells are even capable of engulfing entire unicellular microorganisms. You might have correctly hypothesized that the uptake and release of large particles by the cell requires energy. A large particle, however, cannot pass through the membrane, even with energy supplied by the cell.
8.4.1 Endocytosis
Endocytosis is a type of active transport that moves particles, such as large molecules, parts of cells, and even whole cells, into a cell. There are different variations of endocytosis, but all share a common characteristic: The plasma membrane of the cell invaginates, forming a pocket around the target particle. The pocket pinches off, resulting in the particle being contained in a newly created intracellular vesicle formed from the plasma membrane. The three types of endocytosis are phagocytosis, pinocytosis, and receptor-mediated endocytosis.
Phagocytosis
Phagocytosis (“cell eating”) is the process by which large particles, such as other cells or relatively large particles, are taken in by a cell. For example, when microorganisms invade the human body, a type of white blood cell called a neutrophil will “eat” the invaders through phagocytosis, surrounding and engulfing the microorganism, which is then destroyed by lysosomes inside the neutrophil ( Figure 8.21 ).

In preparation for phagocytosis, a portion of the inward-facing surface of the plasma membrane becomes coated with a protein called clathrin , which stabilizes this section of the membrane. The coated portion of the membrane then extends from the body of the cell and surrounds the particle, eventually enclosing it. Once the vesicle containing the particle is enclosed within the cell, the clathrin disengages from the membrane and the vesicle merges with a lysosome for the breakdown of the material in the newly formed compartment. When accessible nutrients from the degradation of the vesicular contents have been extracted, the newly formed endosome merges with the plasma membrane and releases its contents into the extracellular fluid. The endosomal membrane again becomes part of the plasma membrane.
Pinocytosis
Through pinocytosis (“cell drinking”), cells take in molecules, including water, which the cell needs from the extracellular fluid. Pinocytosis results in a much smaller vesicle than does phagocytosis, and the vesicle does not need to merge with a lysosome ( Figure 8.22 ).
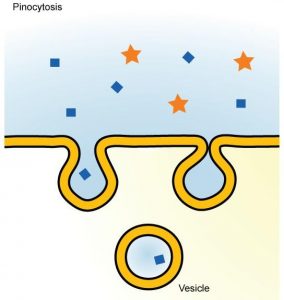
Receptor-mediated Endocytosis
Receptor-mediated endocytosis is a targeted variation of endocytosis that employs receptor proteins in the plasma membrane that have a specific binding affinity for certain substances ( Figure 8.23 ).
Receptor-mediated endocytosis, as in phagocytosis, uses clathrin protein attached to the cytoplasmic side of the plasma membrane. Some human diseases are caused by the failure of receptor-mediated endocytosis. For example, the form of cholesterol termed low-density lipoprotein or LDL (also referred to as “bad” cholesterol) is removed from the blood by receptor-mediated endocytosis. In the human genetic disease familial hypercholesterolemia, the LDL receptors are defective or missing entirely. People with this condition have life-threatening levels of cholesterol in their blood, because their cells cannot clear LDL particles from their blood.
Although receptor-mediated endocytosis is designed to bring specific substances that are normally found in the extracellular fluid into the cell, other substances may gain entry into the cell at the same site. Flu viruses, diphtheria, and cholera toxin all have sites that cross-react with normal receptor-binding sites and gain entry into cells.
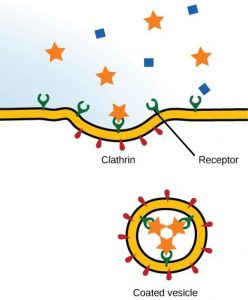
8.4.2 Exocytosis
The reverse process of moving material into a cell is the process of exocytosis . The purpose of exocytosis is to expel material from the cell into the extracellular fluid. Waste material is enveloped in vesicle, which fuses with the interior of the plasma membrane, expelling the waste material into the extracellular space ( Figure 8.24 ). Cells also use exocytosis to secrete proteins such as hormones, neurotransmitters, or parts of the extracellular matrix.
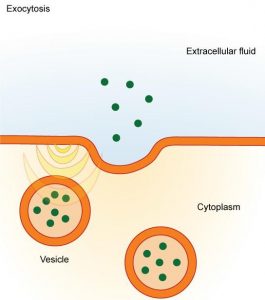
Table 8.2 Methods of transport, energy requirements, and types of material transported
|
|
|
|
| Diffusion | Passive | Small-molecular weight material |
| Osmosis | Passive | Water |
| Facilitated transport/diffusion | Passive | Sodium, potassium, calcium, glucose |
| Primary active transport | Active | Sodium, potassium, calcium |
| Secondary active transport | Active | Amino acids, lactose |
| Phagocytosis | Active | Large macromolecules, whole cells, or cellular structures |
| Pinocytosis and potocytosis | Active | Small molecules (liquids/water) |
| Receptor-mediated endocytosis | Active | Large quantities of macromolecules |
Introduction to Molecular and Cell Biology Copyright © 2020 by Katherine R. Mattaini is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License , except where otherwise noted.
Share This Book
Want to create or adapt books like this? Learn more about how Pressbooks supports open publishing practices.
4.7 Passive Transport
Created by: CK-12/Adapted by Christine Miller
Letting in the Light
Look at the big windows in this house (Figure 4.7.1). Imagine all the light they must let in on a sunny day. Now imagine living in a house that has walls without any windows or doors. Nothing could enter or leave. Or imagine living in a house with holes in the walls instead of windows and doors. Things could enter or leave, but you couldn’t control what came in or went out. Only when a house has walls with windows and doors that can be opened or closed, can you control what enters or leaves. Windows and doors allow you to let in light and the family dog and keep out rain and bugs, for example.
Transport Across Membranes
If a cell were a house, the plasma membrane would be walls with windows and doors. Moving things in and out of the cell is an important function of the plasma membrane. It controls everything that enters and leaves the cell. There are two basic ways that substances can cross the plasma membrane: passive transport — which requires no energy expenditure by the cell — and active transport — which requires energy from the cell.
Transport Without Energy Expenditure By The Cell
Passive transport occurs when substances cross the plasma membrane without any input of energy from the cell. No energy is required because the substances are moving from an area where they have a higher concentration to an area where they have a lower concentration. Concentration refers to the number of particles of a substance per unit of volume. The more particles of a substance in a given volume, the higher the concentration. A substance always moves from an area where it is more concentrated to an area where it is less concentrated.
There are several different types of passive transport, including simple diffusion , osmosis , and facilitated diffusion . Each type is described below.
Simple Diffusion
Diffusion is the movement of a substance due to a difference in concentration. It happens without any help from other molecules. The substance simply moves from the area where it is more concentrated to the area where it is less concentrated. Picture someone spraying perfume in the corner of a room. Do the perfume molecules stay in the corner? No, they spread out, or diffuse throughout the room until they are evenly spread out. Figure 4.7.2 shows how diffusion works across a cell membrane . Substances that can squeeze between the lipid molecules in the plasma membrane by simple diffusion are generally very small, hydrophobic molecules, such as molecules of oxygen and carbon dioxide.
Osmosis is a special type of diffusion — the diffusion of water molecules across a membrane. Like other molecules, water moves from an area of higher concentration to an area of lower concentration. Water moves in or out of a cell until its concentration is the same on both sides of the plasma membrane. In Figure 4.7.3, the dotted red line shows a semi-permeable membrane. In the first beaker, there is an uneven concentration of solutes on either side of the membrane, but the solute cannot cross — diffusion of the solute can’t occur. In this case, water will move to even out the concentration as has happened on the beaker on the right side. The water levels are uneven, but the process of osmosis has evened out the concentration gradient.
Facilitated Diffusion
Water and many other substances cannot simply diffuse across a membrane. Hydrophilic molecules, charged ions, and relatively large molecules (such as glucose) all need help with diffusion . This help comes from special proteins in the membrane known as transport proteins . Diffusion with the help of transport proteins is called facilitated diffusion . There are several types of transport proteins, including channel proteins and carrier proteins. Both are shown in Figure 4.7.4.
- Channel proteins form pores (or tiny holes) in the membrane. This allows water molecules and small ions to pass through the membrane without coming into contact with the hydrophobic tails of the lipid molecules in the interior of the membrane.
- Carrier proteins bind with specific ions or molecules. In doing so, they change shape. As carrier proteins change shape, they carry the ions or molecules across the membrane.
Transport and Homeostasis
For a cell to function normally, the inside of it must maintain a stable state. The concentrations of salts, nutrients, and other substances must be kept within certain ranges. The state in which stable conditions are maintained inside a cell (or an entire organism) is called homeostasis . Homeostasis requires constant adjustments, because conditions are always changing both inside and outside the cell. The transport of substances into and out of cells as described in this section plays an important role in homeostasis. By allowing the movement of substances into and out of cells, transport keeps conditions within normal ranges inside the cells and throughout the organism as a whole.
Watch this video “Cell Transport,” by the Amoeba Sisters:
Cell Transport with the Amoeba Sisters, 2016.
4.7 Summary
- Controlling the movement of things in and out of the cell is an important function of the plasma membrane . There are two basic ways that substances can cross the plasma membrane: passive transport — which requires no energy expenditure by the cell — and active transport — which requires energy.
- No energy is needed from the cell for passive transport because it occurs when substances move naturally from an area of higher concentration to an area of lower concentration.
- Simple diffusion is the movement of a substance due to differences in concentration. It happens without any help from other molecules. This is how very small, hydrophobic molecules (such as oxygen and carbon dioxide) enter and leave the cell.
- Osmosis is the diffusion of water molecules across a membrane. Water moves in or out of a cell by osmosis until its concentration is the same on both sides of the plasma membrane.
- Facilitated diffusion is the movement of a substance across a membrane due to differences in concentration, but it only occurs with the help of transport proteins (such as channel proteins or carrier proteins) in the membrane. This is how large or hydrophilic molecules and charged ions enter and leave the cell.
- Processes of passive transport play important roles in homeostasis . By allowing the movement of substances into and out of the cell, they keep conditions within normal ranges inside the cell and the organism as a whole.
4.7 Review Questions
- What is the main difference between passive and active transport?
- Summarize three different ways that passive transport can occur. Give an example of a substance that is transported in each way.
- Explain how transport across the plasma membrane is related to homeostasis of the cell.
- In general, why can only very small, hydrophobic molecules cross the cell membrane by simple diffusion?
- Explain how facilitated diffusion assists with osmosis in cells. Define osmosis and facilitated diffusion in your answer.
- Can the glucose simply diffuse across the cell membrane? Why or why not?
- Assuming that there are glucose transport proteins in the cell membrane, which way would glucose flow — into or out of the cell? Explain your answer.
- If the concentration of glucose was equal inside and outside of the cell, do you think there would be a net flow of glucose across the cell membrane in one direction or the other? Explain your answer.
- What are the similarities and differences between channel proteins and carrier proteins?
4.7 Explore More
Osmosis and Water Potential, Amoeba Sisters, 2018.
Structure Of The Cell Membrane – Active and Passive Transport, Professor Dave Explains, 2016.
Attributions
Figure 4.7.1
Windows/ The Oyster Suite in Eureka, CA by Drew Coffman on Unsplash is used under the Unsplash License https://unsplash.com/license).
Figure 4.7.2
Diffusion/ Scheme simple diffusion in cell membrane by Mariana Ruiz Villarreal [ LadyofHats] is released into the public domain (https://en.wikipedia.org/wiki/Public_domain).
Figure 4.7.3
Osmosis by OpenStax on Wikimedia Commons is used under a CC BY 3.0 (https://creativecommons.org/licenses/by/3.0) license.
Figure 4.7.4
Scheme facilitated diffusion in cell membrane by Mariana Ruiz Villarreal [ LadyofHats] is released into the public domain (https://en.wikipedia.org/wiki/Public_domain).
Amoeba Sisters. (2016, June 24). Cell transport. YouTube. https://www.youtube.com/watch?v=Ptmlvtei8hw&feature=youtu.be
Amoeba Sisters. (2018, June 27). Osmosis and water potential. YouTube. https://www.youtube.com/watch?v=L-osEc07vMs&feature=youtu.be
Betts, J. G., Young, K.A., Wise, J.A., Johnson, E., Poe, B., Kruse, D.H., Korol, O., Johnson, J.E., Womble, M., DeSaix, P. (2013, April 25). Figure 3.7 Osmosis [digital image]. In Anatomy and Physiology . OpenStax. https://openstax.org/books/anatomy-and-physiology/pages/3-1-the-cell-membrane
Professor Dave Explains. (2016, September 5). Structure of the cell membrane – Active and passive transport. https://www.youtube.com/watch?v=AcrqIxt8am8&feature=youtu.be
A semi-permeable lipid bilayer that separates the interior of all cells from their surroundings.
a type of movement of substances across the cell membrane which does not require energy because the substances are moving with the concentration gradient (from high to low concentration).
The movement of ions or molecules across a cell membrane into a region of higher concentration, assisted by enzymes and requiring energy.
The ability to do work.
The amount of particles of a substance in a given amount of solution.
The movement of a substance from an area of high concentration to an area of low concentration.
The movement of water or other solvent through a plasma membrane from a region of low solute concentration to a region of high solute concentration.
The passive movement of molecules across the cell membrane with the aid of a membrane protein.
The semipermeable membrane surrounding the cytoplasm of a cell.
Attracted to water.
A membrane protein involved in the movement of ions, small molecules, or macromolecules, such as another protein, across a biological membrane.
The smallest unit of life, consisting of at least a membrane, cytoplasm, and genetic material.
The ability of an organism to maintain constant internal conditions despite external changes.
Repelled by water.
Human Biology Copyright © 2020 by Christine Miller is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License , except where otherwise noted.
Share This Book
IMAGES
VIDEO
COMMENTS
Case Study - Cellular Transport Passive Transport vs. Active Transport: When materials need to be transported across the cell membrane, either into or out of the cell, cellular transport occurs. When molecules are moved from a high to low concentration across the membrane, this process is called passive transport because no energy is used.
1. Introduction. Life depends on a membrane's ability to precisely control the level of solutes in the aqueous compartments, inside and outside, bathing the membrane. The membrane determines what solutes enter and leave a cell. Transmembrane transport is controlled by complex interactions between membrane lipids, proteins, and carbohydrates.
If you're seeing this message, it means we're having trouble loading external resources on our website. If you're behind a web filter, please make sure that the domains *.kastatic.org and *.kasandbox.org are unblocked.
Case Study: After running a marathon, an individual is brought into the ER with severe hyponatremia. Hyponatremia occurs when a severely dehydrated person rapidly consumes large amounts of water. This causes the amount of sodium in the blood to drop drastically and the amount of water to greatly increase (blood becomes hypotonic).
Membrane transport proteins therefore play essential roles in cellular function: their actions underpin nerve impulses, muscle contraction, the heartbeat, urine formation and absorption of food in the gut. Perhaps unsurprisingly, inadequate or excess activity of these proteins leads to disease, while a number of common drugs exert their actions ...
In this study, we developed a CRISPRi/a screening strategy to systematically identify nutrient transporters in cells, focusing on characterizing amino acid transport in the K562 leukaemia cell line.
Case Study: Membranes Peter Freddolino and Amy Shih April 13, 2006 1 Introduction to Lipids and Membranes Membranes are essential to cellular organisms. They are like fortresses in that they provide a barrier between the inside and outside with guarded ... transport or digest cellular products and waste. For example, cell organelles like the ...
The case study, "Coat Proteins and Vesicle Transport" (Scales SJ, Pepperkok R, Kreis TE. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COP I. Cell 1997; 90: 1137-1148), examines the role of COPI and COPII in protein transport from the RER to the Golgi complex.
Learn about the structure and functions of the plasma membrane, the cell's boundary that defines its identity and interactions. Explore the mechanisms of passive and active transport, and how they enable cells to exchange materials across the membrane.
A team from UNIGE and UZH has developed a new technique to study membrane transporters in their native environment: the cell. They use nanobodies and electron spin resonance spectroscopy to measure the distance between two magnetic probes attached to the transporter.
The processes of passive and active transport to move substances into and out of cells and help maintain homeostasis. How organisms obtain the energy needed for life, including how the sugar glucose is broken down to produce ATP through the processes of anaerobic and aerobic cellular respiration. The phases of the cell cycle, how cells divide ...
4.7 Summary. Controlling the movement of things in and out of the cell is an important function of the plasma membrane. There are two basic ways that substances can cross the plasma membrane: passive transport — which requires no energy expenditure by the cell — and active transport — which requires energy.
case study - cellular transport - Free download as Word Doc (.doc), PDF File (.pdf), Text File (.txt) or read online for free. Molly visited her doctor complaining of diarrhea after returning from a trip to Africa. Tests revealed that Molly's stool sample had higher than normal salt concentrations but normal sugar levels. A microscope image of Molly's intestinal cells showed active transport ...
Cellular_Transport_Study_Guide -answer key - Free download as Word Doc (.doc), PDF File (.pdf), Text File (.txt) or read online for free. This document provides a review of key concepts in cellular transport: 1) It defines active and passive transport, diffusion, osmosis, equilibrium, and the specific processes of endocytosis, exocytosis, and facilitated diffusion.
Through this worksheet, students will read six unique case studies where cell transport has gone wrong. They will analyze what type of transport is involved in each situation and answer text dependent questions for some. This is a great activity to relate a seemingly boring topic like cell transport to everyday life. Total Pages. 6 pages.