

UK Clinical Research Network Study Portfolio UK Clinical Research Network Study Portfolio
The UKCRN Portfolio comprises four smaller portfolios of studies belonging to the four nations of the United Kingdom. A second version of the search tool is accessible from the UKCRN Search tool and allows searches within the specific portfolios of these countries. The links to this are shown in Figure 1 below and a separate User Guide for this system is available from the UKCRN web site.
Accessibility Statement (This link opens in a new window)
Clinical Governance
Nihr clinical research network (crn) portfolio.
Adoption onto the portfolio has a number of benefits for researchers, such as help in identifying potential research sites, access to patients and the public to carry out ‘ PPI ‘ and advice on recruitment strategy at any point during the study. The CRN offers support to researchers via their Study Support Service and likewise via each portfolio manager and their team.
You can see a breakdown of each portfolio here on our local Clinical Research Network’s page (CRN Wessex).
Portfolio adoption is usually vital to participating NHS Trusts when considering the research studies they wish to undertake, as they are reimbursed for the resource given to conduct the study (e.g. research nurse support, data manager time).
Non-commercial studies that do not require Health Research Authority Approval (i.e. those taking place in public health and social care settings) should apply for CRN support via the Non-commercial Portfolio Application service in CPMS.
In order to use the Non-commercial Portfolio Application service, you must first create an account in CPMS. Find out how to create an account and log in to CPMS here .
Before starting your application, it is expected that you will have:
- Discussed your study with your local CRN (Wessex), through the NIHR early contact and engagement service
- Confirmed the sponsorship arrangements for your study
- Secured full research funding for your study, in line with the Department of Health and Social Care’s AcoRD policy
You should submit your application as soon as you have secured full research funding for your study, in line with the Department of Health and Social Care’s (DHSC) Attributing the costs of health and social care research (AcoRD) policy . There is no need to wait until you are ready to apply for ethical and regulatory approvals.
To complete your application you will need to provide a copy of your study protocol and evidence of the research funding you have secured so have these ready before starting your application.
Once we receive your application it will be reviewed against the Department of Health and Social Care’s Eligibility Criteria (.PDF) , and you will be notified of the outcome via email.
Further guidance on how to apply for NIHR Clinical Research Network (CRN) support through the Non-commercial Portfolio Application service is available on NIHR Learn . A supporting video is also available on the NIHR Youtube channel .
Requirements
In order to be eligible for portfolio adoption, there are three criteria a study must meet:
- The study must be ‘research’ (this is stipulated, as often what’s classed as research outside the NHS setting, is sometimes a service evaluation, quality improvement etc. within the NHS – see this table );
- Have appropriate ethics approval; and Health Research Authority (HRA) Approval where required;
- Have full research funding * – studies that are automatically eligible will have been awarded via open competition and by the NIHR, other areas of central Government, or an NIHR non-commercial partner (for which there is a list ). If the study has received support from multiple funders, then it will be still considered automatically eligible, if one of the funding streams is the NIHR, an area of central Government or a non-commercial partner (found on the list ).
*Other non-commercial funding streams (charities for example) may make your project eligible for the portfolio – check with the portfolio team .
You can read more about study eligibility here , including research funded by overseas partners. There is also a list of FAQs here .
The Portfolio and BU
The source of research funding is the principal determinant of eligibility for NIHR CRN support and so it is encouraged that researchers seek external funding where possible and appropriate.
The amount of funding doesn’t need to substantial in order to be eligible.

Search form
Ukcrn portfolio database.
The National Institute for Health Research Clinical Research Network Coordinating Centre (NIHR CRN CC) has developed the Central Portfolio Management System (CPMS) Portfolio Database . The aim is to build a complete picture of the clinical research taking place across the UK in order to manage the allocation of NHS infrastructure funding.
The database holds records of all research ongoing in the UK that is eligible for the UKCRN Portfolio. In Scotland, studies funded by eligible funders are entered. NHS R&D offices and Scottish Clinical Research Networks are responsible for creating records on the system, however it is the responsibility of Chief Investigators to ensure that their study record is complete and accurate.
Only those studies that are part of the portfolio will have access to infrastructure support. It is therefore, crucial that all eligible studies are included.
This website has now been archived. Please visit us at

Is my research eligible for NIHR portfolio adoption?
For a study to be considered for portfolio adoption it must:
Meet the definition of research (as outlined in the Department of Health and Social Care eligibility criteria ) as follows:
Research can be defined as the attempt to derive generalisable or transferable new knowledge to answer or refine relevant questions with scientifically sound methods. This excludes: audit; needs assessments; quality improvement and other local service evaluations. It also excludes routine banking of biological samples or data except where this activity is integral to a self-contained research project designed to test a clear hypothesis.
Have appropriate ethical approval; and HRA Approval where required.
Have full research funding (i.e. funding to meet all Research Costs in compliance with the AcoRD guidance ).
The source of research funding is the principal determinant of eligibility for NIHR portfolio adoption:
Studies that are funded by the NIHR and / or other areas of central Government and those which are funded by NIHR non-commercial partners, are automatically eligible for consideration for NIHR CRN support provided they meet the definition of research above. Please find more information on the Eligibility for NIHR Clinical Research Network support web page.
How to apply for NIHR CRN support?
If you think your study is eligible for portfolio adoption, please follow the instructions on how to apply for NIHR CRN support . Early engagement with NIHR CRN support helps with optimising study delivery upfront. If it is not automatically eligible, the CRN Eligibility Team and speciality subject specific experts will carry out the assessment and decide whether the study is eligible for portfolio adoption.
If your study is an English-led CTIMP and you are applying for HRA Approval through the HRA and MHRA’s combined review service , you must apply for CRN support through the new Non-commercial Portfolio Application service in CPMS .
The new Non-commercial Portfolio Application service : The following study types can now apply for CRN support via the new service, which allows investigators to apply earlier and receive an eligibility decision sooner to benefit from the full range of support that our study support service offers:
English-led CTIMP (clinical trials of investigational medicinal products) studies which are led in England and going through the Health Research Authority (HRA) and Medicines and Healthcare products Regulatory Agency’s (MHRA) Combined Review Service , and;
English-led studies that do not need, and therefore are not applying for, HRA Approval in the Integrated Research Application System (IRAS) .
Studies that require HRA Approval but are not being progressed through combined review: These studies should continue to apply for NIHR CRN support by selecting ‘yes’ to question 5b of the IRAS Project Filter. This will ensure key information from your IRAS submission is automatically shared with us. This information, including the IRAS form, the study protocol and grant award letter(s), will be used to determine eligibility. You will be notified of the outcome, via email.
If you are unable to apply via either of these routes, contact your Local CRN for advice on how to proceed.
If you require any local advice on applying for portfolio adoption or on eligibility criteria please contact [email protected]
Once your study has been granted the portfolio eligibility status, you can contact the NIHR study delivery officer for local access to CRN-funded possibilities to aid recruitment.
Further information
Further information on eligibility for NIHR support can be found in eligibility FAQs .
You can read about the benefits of portfolio adoption here .
- Store finder
Registering with the portfolio
The UK Clinical Research Network (UKCRN) helps to provide the infrastructure that allows high-quality clinical research to take place in the NHS so that patients can benefit from new and better treatments.
The UK Clinical Research Network
The UKCRN consists of the National Institute for Health Research Clinical Research Network (NIHR CRN) in England and its equivalents in the devolved nations. Through the UKCRN, researchers can have access to support at the planning, set-up and delivery phases of a study, including advice and support to overcome barriers to successful recruitment.
The UKCRN portfolio database
The UKCRN portfolio database is a database of high-quality clinical research studies that make up the clinical study portfolios of England (the NIHR CRN portfolio ), Northern Ireland, Scotland and Wales. Studies accepted on to the UKCRN portfolio are eligible for consideration for research infrastructure and service support from the NIHR CRN in England and from equivalent structures in the devolved nations. The portfolio is also used to monitor the progress of clinical studies by regularly recording recruitment data.
We are an NIHR non-commercial partner and therefore any study funded by us is automatically eligible for inclusion in the UKCRN portfolio. We receive regular reports from the portfolio database charting progress with recruitment into the studies we support, and we use these reports to monitor the progress and outputs of the award.
Condition of award
If you are awarded funding for a clinical study that requires support from the NIHR CRN or its equivalents, as a condition of award, we require that you:
- Ensure your clinical study is registered on the portfolio.
- Ensure your grant reference number is recorded on the portfolio.
- Upload recruitment data on a regular (monthly) basis onto the portfolio database.
Visit the NIHR CRN website to find out ways of accessing advice and support and how to upload recruitment data.
https://www.gosh.nhs.uk/our-research/our-research-infrastructure/joint-research-and-development-office-rd/research-management-and-governance-team-rmg-team/nihr-crn-portfolio/
NIHR CRN Portfolio
The NIHR Clinical Research Network (CRN) Portfolio is the group of high-quality clinical research studies that have satisfied certain eligibility criteria. The NIHR CRN Portfolio is part of the UK Clinical Research Network Portfolio. Important things to remember about the NIHR CRN Portfolio:
- If you want to submit your research project for adoption to the Portfolio, please check ‘yes’ to either question 5a or 5b in the IRAS form project filter and complete and submit the IRAS Portfolio Adoption Form (PAF).
- Non-commercial studies that have been adopted on the Portfolio are eligible for consideration for service support costs from the Clinical Research Network in England through CLRN contingency funding.
- Portfolio studies are registered on the UK CRN Portfolio database and monthly recruitment data must be uploaded. The recruitment data informs the allocation of NHS infrastructure for research (including NHS Support Costs)
More information about the Portfolio, including details of eligibility and benefits of inclusion, can be found at the NIHR’s website .
Please also contact [email protected] for further information.
- UK Clinical Research Network (UKCRN)
Clinical Research Network in England
- Clinical Research Networks in Northern Ireland
- Clinical Research Networks in Scotland
- Clinical Research Networks in Wales
- European Context
- Joint Funding Initiatives
- Coordination of Experimental Medicine
- UKCRC Registered Clinical Trials Units
- UKCRC Registered CTUs Resource Finder
- UKCRC Tissue Directory and Coordination Centre
Clinical research infrastructure in England is provided through the NIHR Clinical Research Network.
The Network supports the delivery of a portfolio of clinical research studies, including life-sciences industry studies, across all parts of the NHS in England.
It does this by providing funds to hospitals and surgeries to invest in clinical research nurses, and other clinical staff. This highly-trained workforce matches patients with appropriate study participation opportunities, and carries out the clinical duties required by the studies, enabling research teams to answer the research question to time and target. Network funding also covers costs related to study delivery such as x-rays and scans.
The NIHR Clinical Research Network also works to ensure that clinical research occupies the place it deserves in the day-to-day work of the NHS, by encouraging clinical professionals to engage actively in research activities, for the benefit of patients and the NHS service as a whole.
Click to view full size
The Network comprises 15 areas that, together, cover the whole of England. Each area is responsible for delivery of a portfolio of studies spanning 30 specialities. These specialities are: Ageing; anaesthesia; perioperative medicine and pain management; cancer; cardiovascular disease; children; critical care; dementias and neurodegeneration (DeNDRon); dermatology; diabetes; ear, nose and throat; gastroenterology; genetics; haematology; health services and delivery research; hepatology; infectious diseases and micrbiology; injuries and emergencies; mental health; metabolic and endocrine disorders; musculosketetal disorders; neurological discorders; opthalmology; oral and dental health; primary care; public health; renal disorders; reproductive health and childbirth; respiratory disorders; stroke; surgery.
The Network is managed by a National Coordinating Centre, which takes responsibility for the cross-cutting initiatives to improve study delivery.
Useful Links
- NIHR Clinical Research Network Coordinating Centre
- UKCRN portfolio database: England
- UK-wide working across UKCRC Activities

UCL Queen Square Institute of Neurology

NIHR Portfolio Adoption

The NIHR Portfolio consists of high-quality clinical research studies across a range of over 26 speciality groups that are eligible for consideration for research support from the Clinical Research Network in England. Portfolio adoption can provide management of current studies, facilitate feasibility of future studies and support with staffing. Activity data of studies and recruitment from the NIHR portfolio is used to inform NHS infrastructure allocation and supports the performance management of each of the Clinical Research Networks. The QSCTC encourages researchers to adopt their studies onto the NIHR portfolio providing that they meet the eligibility criteria. Divisional Portfolio Officers are available as a single point of contact to guide research through the adoption process, help with study feasibility and manage performance. The Portfolio officer working with the QSCTC is Amal Qureshi ( [email protected] ).
Benefits of Portfolio adoption:
- Access to a local network of research support staff such as research nurses and other healthcare professionals
- Offering support to ensure research studies are feasible to be carried out under the NHS
- Providing access to staff that can carry out local governance checks and obtain NHS permission
- Managing study performance by monitoring Key Performance Indicators such as time to target recruitment in studies
To access more information on eligibility, when and how to apply open the following links:
Eligibility Criteria
Research studies funded by the NIHR and NIHR Partners are automatically eligible for portfolio adoption. Otherwise, studies need to have a source of funding from open competition. Also, the research being carried out has to be of value to the NHS and needs to be feasible within the realities of the NHS.
- Further information of portfolio eligibility
When to apply
As long as your research study meets the eligibility criteria, it is important to apply for portfolio adoption at an early stage (i.e. before completion of the R&D form and SSI form). This gives enough time for the research study to be processed through the Coordinated System for gaining NHS Permission (CSP) system for R&D (NHS) approval and potential funding being received from the network.
How to apply
When completing the IRAS form, Question 5b states ‘Do you want your NHS R&D application to be processed through the NIHR Coordinated System for gaining NHS Permission’ –for this select Yes. This will then generate a portfolio adoption form (PAF) through IRAS where North Thames will be the local clinical research network for Queen Square, and you will be notified once your study is adopted onto the NIHR portfolio.
- Applying for Portfolio Adoption

Contact Details
Queen Square Clinical Trial Centre UCL Institute of Neurology 7 Queen Square 5th Floor London WC1N 3BG Telephone: 020 3448 3555 E-mail: [email protected]
Useful Links:
NIHR Clinical Research Network Website
Attributing the costs of health and social care Research & Development (AcoRD) guidelines
Joint Research office
UCL Clinical Trials Unit
- Portfolio Maps
- Breast Group
- Strategic priorities
The NCRI Group portfolio maps are a visual overview of the UK cancer clinical studies within the NIHR CRN portfolio, which are preparing to open for recruitment (‘In set-up’) or are actively recruiting patients (‘Open’). The maps categorise studies by disease site, research area and Local Clinical Research Network (LCRN). When sharing or adapting these maps, please cite NCRI Portfolio Maps.
More from NCRI
The small study for treating early-stage breast cancer.
The SMALL study is a phase 3 clinical trial, receiving over £2.3M in funding from the NIHR HTA, designed to test if radiotherapy-guided surgical removal…
Identifying more harm than good: The PRIMETIME breast cancer study
PRIMETIME is a research study in early breast cancer investigating whether it’s possible to identify a group of women with a very low risk of…
Analyses of genetic variants show the effect of sleep on breast cancer risk
Women who are “larks”, functioning better at the beginning of the day than the end of the day, have a lower of risk breast cancer,…
Read more here
Portfolio Maps
_________________________
The Clinical Research Network West Midlands (CRN WM) Digital Portfolio Maps have been developed to inform Researchers and Healthcare Professionals of the research studies available locally (West Midlands) and nationally.
The CRN WM Digital Portfolio Maps aim to:

Act as a central hub for accessing clinical trials information relating primarily to the West Midlands

Provide a visual aid with the ability to filter research studies by main specialty, sub specialty, study title, keyword search, study identification number, Hospital Trust, study type, and study status

Support the monitoring and management of research studies to help ensure there is a balanced portfolio

Support local partner organisations to aid patient referrals into research studies

Encourage collaboration and sharing of best practice between researchers

Provide accessibility of the portfolio across all formats of technology
Studies in the West Midlands
____________________
The West Midlands Studies page details the research studies that are open and in set-up in the Clinical Research Network West Midlands. Each study record includes the following information; Main specialty, study name, NIHR portfolio identification number, study title, study status, commercial/non-commercial and study wide recruitment percentage rate.
Additional information can be accessed by clicking on the study record, this will show the study timeframe & target, inclusion/exclusion criteria, participating Hospital Trusts in the West Midlands. L inks are included for the Public Open Data Platform (ODP), Be Part of Research (BP o R) (for commercial studies), Central Portfolio Management System (CPMS), EDGE (Local Portfolio Management System) and study website. The Public ODP and Be Part of Research is accessible to anyone, however logins are required for CPMS and EDGE.
Please note that commercial portfolio studies (open) are only included if they are accessible to the public on the Be Part of Research website.

See all NIHR studies
This NIHR Studies page details the NIHR portfolio studies that are open and in set-up. Each study record details the main specialty, study name, NIHR portfolio identification number, study title, study status and study wide recruitment percentage rate.
Additional information can be viewed by clicking on the study record, this will show the study timeframe & target, inclusion/exclusion criteria etc. Links are included for the Public Open Data Platform (ODP), Be Part of Research (BP o R) (for commercial studies), Central Portfolio Management System (CPMS), EDGE (Local Portfolio Management System) and study website.
The Public ODP and Be Part of Research is accessible to anyone, however logins are required for CPMS and EDGE.
The information and data included in the Portfolio Maps has been taken from the NIHR Central Portfolio Management System, EDGE (Local Portfolio Management System) and Be Part of Research.
This site is best viewed in Google Chrome, IE11, Mozilla Firefox, Opera and Safari browsers.
The Digital Portfolio Maps are adapted from initial concepts developed by the National Cancer Research Institute.
© 2016 developed and designed exclusively by the Clinical Research Network West Midlands, hosted by The Royal Wolverhampton NHS Trust .
Log in using your username and password
- Search More Search for this keyword Advanced search
- Latest content
- Current issue
- For authors
- BMJ Journals
You are here
- Volume 102, Issue 8
- NIHR Clinical Research Networks: what they do and how they help paediatric research
- Article Text
- Article info
- Citation Tools
- Rapid Responses
- Article metrics
- Hanna Lythgoe 1 , 2 ,
- Victoria Price 1 , 2 ,
- Vanessa Poustie 3 ,
- Sabah Attar 3 ,
- Daniel Hawcutt 1 , 2 ,
- Jennifer Preston 2 ,
- Michael W Beresford 1 , 2 , 3
- 1 Department of Women's and Children's Health , Institute of Translational Medicine, University of Liverpool , Liverpool , UK
- 2 NIHR Alder Hey Clinical Research Facility, Alder Hey Children's NHS Foundation Trust , Liverpool , UK
- 3 NIHR CRN: Children, Institute of Translational Medicine, University of Liverpool , Liverpool , UK
- Correspondence to Professor Michael W Beresford, Institute in the Park, Alder Hey Children's Hospital, Eaton Road, Liverpool L12 2AP, UK; M.w.beresford{at}liverpool.ac.uk
This review provides paediatricians with an update on the new structure of the National Institute for Health Research's (NIHR) Clinical Research Network (CRN): Children and its role within the wider NIHR infrastructure. The network supports delivery of high-quality research within the NHS in England and supports researchers, through provision of staff and resources, with feasibility, site set-up, patient recruitment and study management. Since 2013, over 80% of commercial contract studies running within the UK sat within the UKCRN Portfolio. Of the diverse, increasing portfolio of studies supported by the network, many studies are interventional, with 33% being randomised controlled studies. Recruitment to studies supported by the network through the Children's Portfolio has consistently improved. Over 200 000 participants have been recruited to the Children's Portfolio studies to date, and there are currently approximately 500 studies open to recruitment. The CRN: Children has successfully involved patients and the public in all aspects of study design and delivery, including through the work of Generation R. Challenges remain in conducting paediatric research and the network is committed to supporting Children's research and further building on its achievements to date. Education and engagement of paediatricians within the network and research is important to further improving quality and delivery of paediatric research.
- Clinical Research Networks
- Child Health
- Paediatrics
- Clinical Trials
- National Institute for Health Research
https://doi.org/10.1136/archdischild-2016-311057
Statistics from Altmetric.com
Request permissions.
If you wish to reuse any or all of this article please use the link below which will take you to the Copyright Clearance Center’s RightsLink service. You will be able to get a quick price and instant permission to reuse the content in many different ways.
HL and VP contributed equally.
Contributors We can confirm that all authors meet the ICMJE recommendations for authorship.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Read the full text or download the PDF:
This site is optimised for modern browsers. For the best experience, please use Google Chrome, Mozilla Firefox, or Microsoft Edge.
CRN West of England

Quick Links
- Health and Care Professionals
- Researchers
- Patients, Carers and the Public
The Clinical Research Network (CRN) West of England, hosted at University Hospitals Bristol and Weston NHS Foundation Trust, facilitates research in NHS, Public Health and social care settings, supporting portfolio studies in 31 specialties in sites such as universities, schools, care homes, hospices, prisons and clinical settings.
Latest news about West of England
News: 77% of GP practices in the West of England took part in research

News: Volunteers needed for Mpox vaccine study in Bristol
News: Consultant Medical Ophthalmologist in West of England recognised for outstanding contributions to research

Latest case studies from West of England
Case study: Principal Investigator (PI) and Assistant Principal Investigator (API) encourage both health professionals and study leads to register for NIHR API scheme

Case study: What does effective research leadership look like? Insights from our Research Scholars’ Programme

Case study: New mum shares experience of taking part in research study for premature babies

We are funded by the Department of Health and Social Care to facilitate the delivery of research studies, by supporting our partner organisations in the West of England.
Our partner organisations comprise NHS trusts, primary care, public health and social care.
We work together to ensure that research studies that are included on the NIHR CRN Portfolio are set up quickly and are delivered to time and to target.
We facilitate both commercial and non-commercial research. We help locally hosted studies to secure sites across the country and we also help to attract studies to the West of England.
What area does the West of England cover?
Our area comprises the City of Bristol, Bath and North East Somerset (BaNES), Swindon, Gloucestershire, North Somerset and part of Wiltshire.
In our area we work with seven Trusts:
- University Hospitals Bristol and Weston NHS Foundation Trust
- North Bristol NHS Trust
- Royal United Hospitals Bath NHS Foundation Trust
- Gloucestershire Hospitals NHS Foundation Trust
- Avon and Wiltshire Mental Health Partnership NHS Trust
- Gloucestershire Health and Care NHS Foundation Trust
- Great Western Hospitals NHS Foundation Trust, Swindon
We work in approximately 100 primary care settings, both single sites and collaborations, across three Integrated Care Systems (ICSs).
- Bath and North East Somerset, Swindon and Wiltshire
- Bristol, North Somerset and South Gloucestershire
- Gloucestershire
What support can the Clinical Research Network offer?
If a study is on the Portfolio, we can offer:
- support for a study from the outset via the Study Support Service
- funding for study support as appropriate, to ensure good quality research opportunities for people in the West of England
- training for staff (for example Good Clinical Practice, Informed Consent and the Principal Investigator Masterclass)
- specialist advice from research and specialty experts .
- the sharing of effective research strategies locally, nationwide and across specialties.
We also have a peripatetic team, comprising Research Nurses and Clinical Research Practitioners (CRPs), who are trained to offer support with the delivery of studies (depending on availability) in a wide range of settings such as hospices, schools and care homes.
Our ENRICH network brings together care home staff, residents and their families with researchers. It provides a toolkit of resources to help care homes make the most of research and researchers to set up and run studies effectively and collaboratively in care homes. Your local ENRICH contact can be found on the ENRICH website.
We also support Join Dementia Research , a national service that enables you to register your interest and be matched with suitable research studies.
Our promotional work increases opportunities for the public to take part in clinical research and seeks to embed research as a standard component of care, for patients and carers, healthcare professionals and NHS decision makers.
What is the Portfolio?
The CRN Portfolio is the collection of studies that qualify for CRN support. To receive CRN support, your study must be on the Portfolio and to be on the Portfolio your study must be eligible for inclusion. Find more information about the eligibility criteria .
How can I find out if a study is eligible?
Please email [email protected] in the first instance and our team will get back to you.
Meet our new Alliance member! NIHR's Clinical Research Network
4 August 2020
Guest Blog: Stephen Lock, Interim Chief Information and Technology Officer, NIHR CRN
Share this page
The National Institute for Health Research Clinical Research Network (NIHR CRN) is delighted to be joining the UK Health Data Research Alliance . Our unique data and expertise will make an important contribution in the collective effort to improve clinical research across the nation. To explain how this can happen, we need first to explain a little about the data we hold and the work we do.
The NIHR CRN maintains data in order to help coordinate a national portfolio of clinical research. The datasets we hold are used to manage core study information, capacity and capability data, study site information, research activity data and research management information. In addition, the NIHR’s annual research statistics – published at the beginning of each financial year – provide the most comprehensive data around the state of health research across England.
Our Central Portfolio Management System (CPMS) holds 10-years worth of data on more than 26,000 studies that have recruited in excess of 9 million participants. We are able to analyse this data by, for example, location, site type, speciality, phase, funder type, study design, geographical scope and funder type.
More recently, we’ve had great success in linking this data to other datasets. This has enabled us to, for example, analyse research activity data in relation to disease prevalence (through linking data shared by Public Health England), or admission rates (by incorporating NHS England data). This work has been incredibly helpful in enabling research to be targeted at areas of unmet need – a key priority of the Chief Medical Officer who chairs the NIHR Strategy Board.
The CRN’s Open Data Platform (ODP) is a Business Intelligence (BI) Platform. It is used by more than 3,000 researchers, academics and healthcare professionals to visualise our research data in creative ways. For example, the ODP Research Targeting Tool is now used to help place research studies in areas of high prevalence of disease.
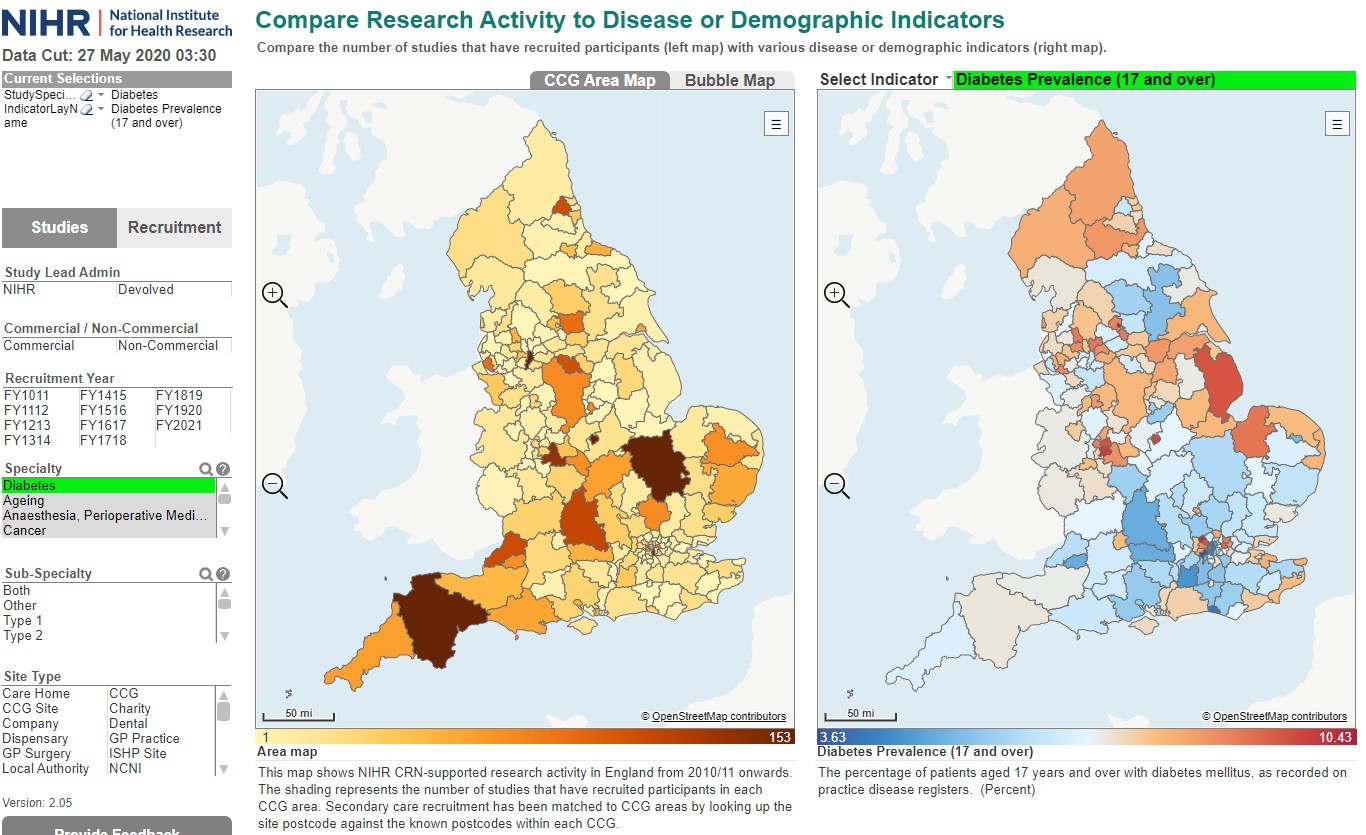
Image from the NIHR’s Open Data Platform, Research Targeting Tool
The CRN also manages the Be Part of Research service which allows people of all ages and backgrounds, from all over the country, to take part and help shape research. Recently the BPoR team has worked closely with NHS Digital to develop the Coronavirus consent to contact service . This new research registry forms a crucial part of the work of the Government’s Vaccines Task Force that will help us accelerate the recruitment of research participants to vital vaccine trials, right across the country.
As an organisation, we realise the power of linking data and organisations together. As a network by design, we have expertise in working across boundaries (every single NHS trust in England is involved in supporting NIHR CRN research) and using data in creative ways to give insight and to aid decision making.
We are certain that by joining the UK Health Data Research Alliance and by opening up our knowledge, network and data further, we can help HDRUK to thrive and really make a difference to people who deserve access to research. We can’t wait to get started.

We couldn’t find any results matching your search.
Please try using other words for your search or explore other sections of the website for relevant information.
We’re sorry, we are currently experiencing some issues, please try again later.
Our team is working diligently to resolve the issue. Thank you for your patience and understanding.
News & Insights

Quest Diagnostics' (DGX) New Study Exposes Alarming Data on STI
August 15, 2024 — 09:05 am EDT
Written by Zacks Equity Research for Zacks ->
Quest Diagnostics DGX , in collaboration with the University of Alabama, conducted a study that suggests adherence to guideline-based laboratory testing and treatment of pregnant women for two of the most prevalent sexually transmitted infections (STIs) is suboptimal in the United States. This could potentially result in dire effects on maternal and newborn health.
The study, titled “Chlamydia and gonorrhea testing in pregnancy: Time to improve adherence and update recommendations”, was published in the peer-reviewed Journal of Lower Genital Tract Disease — the official journal of the American Society for Colposcopy and Cervical Pathology. It is based on deidentified results of lab tests performed in all 50 states and the District of Columbia for more than four million pregnancies.
More on the Study and Findings
Per key findings, more than 4% of women who received guideline-based screening for chlamydia or gonorrhea in pregnancy during the first trimester received a positive result for one or both infections. Among these, more than one in three (35.1% chlamydia and 36.9% gonorrhea) did not receive a follow-up negative test before delivery. This reflects a possibility that they may not have been treated and cured or were treated and cured but then reinfected before birth.
Furthermore, nearly 2% of patients who received a negative test result for chlamydia or gonorrhea early in pregnancy later tested positive, suggesting a persistent risk of infection during pregnancy. Of these patients, about one in two (53% chlamydia and 49.3% gonorrhea) remained positive prior to delivery.
The findings reveal gaps in guideline-based care intended to reduce the risk of infection and medical complications. Untreated chlamydia and gonorrhea can lead to heightened risks of infertility and pelvic inflammatory disease in women. The chance of transmission during birth is approximately 50%, raising the potential for newborns to develop infections of the eye (conjunctivitis), lungs (pneumonia) and other health problems.
While the study benefits from its large size, national representation and use of objective laboratory data, it lacks the clinical follow-up information for the positive cases as testing was limited to one national clinical laboratory. The authors did not evaluate other STIs, such as syphilis. However, they caution that these patterns of irregular adherence to guideline-based testing in maternal care may extend to other conditions.
Conclusions of the Study
The authors of the study believe that reinfections (or ineffectively treated initial infections) may lead to more deliveries while women are positive for one or more STIs, affecting both maternal and newborn health. They also concluded that current guidelines are inconsistent and proposed several recommendations for improvements. For instance, the Centers for Disease Control and Prevention (“CDC”) recommends women be retested for cure at four weeks of pregnancy, while the USPSTF recommends retesting before three weeks.
Additionally, the present guidelines do not recommend screening women after the age of 25 years unless there are risk factors (such as multiple partners) based on much earlier data from 1988.
The guidelines recommend that all women under the age of 25 be screened, regardless of perceived risk.According to the CDC, the cases of sexually transmitted diseases are at an all-time high, with more than 2.5 million cases of syphilis, gonorrhea and chlamydia reported in the United States in 2022.
Industry Prospects
Per a report by Grand View Research, the global STI testing market was valued at $10.1 billion in 2023 and is expected to witness a CAGR of 7.2% by 2030. The market is primarily influenced by advancements in diagnostic technology and increased government efforts to promote early detection and treatment.
Updates From Peers
Within the broader MedTech industry, several key players are involved in STI testing and diagnostics. These companies are also making other notable developments in their respective businesses.
Women’s health-focused company, Hologic HOLX , dominates the U.S. markets for STI testing and is well-positioned to capture a larger share internationally. Last month, it acquired Endomagnetics Ltd (Endomag), the UK-based developer of breast cancer surgery technologies, in a transaction valued at approximately $310 million. The latter’s wireless breast surgery localization and lymphatic tracing solutions, including the Magseed marker, the Magtrace lymphatic tracer and the Sentimag platform, will complement and diversify Hologic’s expanding interventional breast health portfolio.
Renowned for its Sample to Insight solutions, QIAGEN QGEN provides a range of STI tests through its Sexual & Reproductive Health portfolio, including tests for Chlamydia trachomatis and Neisseria gonorrhoeae infections. The company recently collaborated with the University of Montana to drive the implementation of next-generation sequencing and forensic investigative genetic genealogy in identifying human remains.
Furthermore, it introduced 35 new wet-lab-tested digital PCR Microbial DNA Detection Assays for the QIAcuity digital PCR platform. These assays target pathogens responsible for tropical diseases, STIs and urinary tract infections, further solidifying the company’s leadership in microbial detection and analysis.
Another peer, Thermo Fisher Scientific TMO , offers several types of manual, automated or semi-automated in vitro diagnostic STI test kits and other STI testing products. The company’s SeCore CDx HLA A Sequencing System was recently granted the FDA 510(k) clearance as a companion diagnostic for Adaptimmune’s TECELRA (afamitresgene autoleucel) T-cell receptor therapy for unresectable or metastatic synovial sarcoma.
TMO also introduced a novel pre-transplant risk assessment (PTRA) assay that helps assess the risk of early acute rejection in kidney transplant recipients. The One Lambda PTR Assay is the first test of its kind, aiming to help inform clinical decision-making and support more personalized patient management, with the ultimate aim of improving outcomes.
5 Stocks Set to Double
Each was handpicked by a Zacks expert as the #1 favorite stock to gain +100% or more in 2024. While not all picks can be winners, previous recommendations have soared +143.0%, +175.9%, +498.3% and +673.0%.
Most of the stocks in this report are flying under Wall Street radar, which provides a great opportunity to get in on the ground floor.
Want the latest recommendations from Zacks Investment Research? Today, you can download 7 Best Stocks for the Next 30 Days. Click to get this free report
Quest Diagnostics Incorporated (DGX) : Free Stock Analysis Report
Thermo Fisher Scientific Inc. (TMO) : Free Stock Analysis Report
Hologic, Inc. (HOLX) : Free Stock Analysis Report
QIAGEN N.V. (QGEN) : Free Stock Analysis Report
To read this article on Zacks.com click here.
Zacks Investment Research
The views and opinions expressed herein are the views and opinions of the author and do not necessarily reflect those of Nasdaq, Inc.

Stocks mentioned
More related articles.
This data feed is not available at this time.
Sign up for the TradeTalks newsletter to receive your weekly dose of trading news, trends and education. Delivered Wednesdays.
To add symbols:
- Type a symbol or company name. When the symbol you want to add appears, add it to My Quotes by selecting it and pressing Enter/Return.
- Copy and paste multiple symbols separated by spaces.
These symbols will be available throughout the site during your session.
Your symbols have been updated
Edit watchlist.
- Type a symbol or company name. When the symbol you want to add appears, add it to Watchlist by selecting it and pressing Enter/Return.
Opt in to Smart Portfolio
Smart Portfolio is supported by our partner TipRanks. By connecting my portfolio to TipRanks Smart Portfolio I agree to their Terms of Use .
Internet Explorer is no longer supported by Microsoft. To browse the NIHR site please use a modern, secure browser like Google Chrome, Mozilla Firefox, or Microsoft Edge.
Study eligibility for Clinical Research Network Support - FAQs

Published: 01 June 2019
Version: December 2023/2.1
Introduction
The purpose of this document is to provide the NIHR Clinical Research Network with a set of Frequently Asked Questions (FAQs) to support the implementation of the Department of Health and Social Care Eligibility Criteria for NIHR Clinical Research Network Support (September 2023) policy.
Frequently asked questions
E1. my study supports the establishment / running of a tissue bank, a disease registry, data bank, cohort or other resource which underpins a number of research studies. is it eligible for nihr clinical research network support.
A “research project” is defined as a structured activity which is intended to provide new knowledge which is generalisable or transferable. The establishment and running of tissue banks or disease registries does not, in itself, constitute a research project (as there is no “research question”) and these activities are therefore not eligible for Clinical Research Network (CRN) support.
However, activities such as the collection and banking of biological samples, inclusion of patient details on a registry, or development of a patient cohort, which form an integral part of a structured research project, or research projects which utilise such resources, are eligible for CRN support (subject to meeting the other eligibility criteria).
The assessment of whether or not a study meets the DHSC Eligibility Criteria for NIHR CRN Support definition of research is made independently of how the study sponsor has classified the study (as per the UK Policy Framework), for the purposes of gaining ethical approval. The aims and objectives of the work and its potential to generate generalisable and transferable new knowledge should be carefully considered. Studies that have undergone ethical review as tissue banks or databases, but which meet the above definition of research and all other aspects of the Eligibility Criteria, can be supported by the CRN, if the following are satisfied: (1) The research questions and anticipated outcomes are clearly stated; and (2) The research methodology to be used (in addition to methods of sample collection / processing / storage) are clearly described; and (3) The outcome(s) can reliably be extrapolated from the subjects who participated to a broader patient population and a broader range of clinical settings; and (4) Evidence is provided to confirm that that funding secured covers all research costs as well as sample collection / processing / storage.
Please note that whilst studies using previously collected samples or data only, may be included on the Portfolio, if they meet the definition of research and all other requirements of the Eligibility Criteria, they cannot upload recruitment. Recruitment can only be captured when new informed consent is given for the collection of new samples or data (i.e. samples/data that have not previously been stored).
E2. My study supports the undertaking of a local service evaluation/improvements. Is it eligible for NIHR Clinical Research Network support?
No, a “research project” is defined as a structured activity which is intended to provide new knowledge which is generalisable or transferable. The undertaking of a local service evaluation does not, in itself, constitute a research project (as there is no “research question”) and these activities are therefore not eligible for Clinical Research Network (CRN) support. However, if the evaluation (i) involves services delivered in more than one NHS / care organisations (eg. NHS Trust or care home); (ii) can be scaled across other organisations or services; and (iii) the outcome can reliably be extrapolated from the subjects who participated to a broader patient population and a broader range of clinical settings, then this evaluation would be considered eligible (subject to meeting the other eligibility criteria) since the activity is generating generalisable or transferable new knowledge.
E3. My study is funded by the NIHR or other areas of central government. Is it eligible for NIHR Clinical Research Network support?
All funding streams administered by, and research commissioned from, central government sources (eg. NHS England, NICE, Research Councils) or any branch of the NIHR, that are provided for the primary purpose of undertaking research and not intended to support infrastructure (including NHS Service Support Costs), are considered automatically eligible for NIHR CRN support.
Studies funded by regional government awards that are intended to support local, not national, activities, including Academic Health Science Networks are not considered eligible, even where the activity being funded is classified as ‘research’.
E4. My study is funded by a non-commercial (non profit making) organisation (e.g. a research charity). Is it eligible for NIHR Clinical Research Network support?
Non-commercial studies are eligible to receive NIHR Clinical Research Network support (CRN) if (1) the non-commercial organisation / funder is an NIHR Non-commercial Partner and has self-declared that the funding stream supporting the study meets the Eligibility Criteria and (2) the study satisfies all other aspects of the Eligibility Criteria including the definition of research (see section 2.1). NIHR Partners are non-commercial organisations that:
- Award research funds as a result of open national competition across England with high quality peer review; and
- Fund research that is of clear value to the NHS, social care or public health; and
- Take appropriate account of the priorities, needs and realities of the NHS, social care or public health in making decision about the research they fund.
E5. My study is funded by a university, college or local healthcare organisation. Is it eligible for consideration for NIHR Clinical Research Network support?
No, UK universities, colleges and local healthcare organisations (including NHS Trusts) do not fulfil the criteria for NIHR non-commercial Partner status, since funding does not meet the open competition requirement. Studies funded by these organisations are therefore not eligible for NIHR Clinical Research Network support.
E6. My not for profit organisation / charity funds research in England. Can I be considered for NIHR Non-commercial Partner status?
Yes, non-commercial organisations which fund research across England can apply to become an NIHR Non-commercial Partner via a self declaration process. Interested organisations should contact the Study Support Service Helpdesk at [email protected] . By signing the self-declaration, funding organisations are confirming that the funding streams they administer meet the NIHR Non-commercial Partner criteria as set out in the Department of Health and Social Care policy document “Eligibility for NIHR Clinical Research Network Support (September 2023)” , see FAQ E4 for further information.
E7. My study is funded as part of a programme or centre grant. Is it eligible for NIHR Clinical Research Network support?
Individual studies funded as part of programme or centre grants are required to have undergone protocol peer review before they can be considered for NIHR Clinical Research Network support. Appendix 1 of the Eligibility Criteria (September 2023) policy document outlines the standard of peer review required. It is a study Sponsor's responsibility to provide confirmation of appropriate peer review.
E8. My study is funded as part of a Research Training Award. Can the work I do be supported by the NIHR Clinical Research Network?
Individual studies funded as part of Research Training Awards are required to have undergone protocol peer review before they can be considered for Clinical Research Network support. Eligibility for NIHR Clinical Research Network support is determined on a study-by-study basis, with emphasis on the study rather than the activity of an individual. If you hold a Research Training Award and the specific project which you are working on underwent protocol peer review then no further peer review is required. However, if your project was not peer reviewed as part of the grant award process, confirmation that the study has been peer reviewed in line with the Eligibility Criteria will be required from the study sponsor before the study can be considered for Clinical Research Network support. If your personal award is funded as an Investigator Initiated Trial, or by an overseas Government or overseas charity, the study will require formal consideration through the non-commercial extended review process. Confirmation that the specific project has been subject to high quality peer review according to the standards outlined in Appendix 1 of the Eligibility Criteria (September 2023) policy document will be required.
E9. My study is supported by several non-commercial funding organisations. How is eligibility determined?
A non-commercial study supported by multiple funders is automatically eligible for consideration for NIHR Clinical Research Network support if (1) one of the funding streams is administered by the NIHR, other area of central Government or an NIHR Non-commercial Partner that has self-declared the individual stream to be ‘eligible’ and (2) the study satisfies all other aspects of the Eligibility Criteria including the definition of research (see section 2.1).
If none of the funders include the NIHR, other area of Central Government or an NIHR Non-commercial Partner then the study may still be considered for Clinical Research Network support via the non-commercial extended review process.
E10. My non-commercial study is supported by a funding stream which has multiple funding partners. How is eligibility determined?
The eligibility of studies that are supported by a funding stream which has multiple funding partners will be determined by the organisation who has managed the funding competition, specifically the peer review process. For example, the Stroke Association and the British Heart Foundation operated a joint programme grant funding stream. The Stroke Association managed the funding competition, including the peer review process and so it is the Stroke Association who was required to self-declare with respect to this funding stream.
E11. My study is funded by an overseas Government. Is it eligible for NIHR Clinical Research Network support?
Studies that are funded by overseas Governments are considered for eligibility for CRN support via the non-commercial extended review process. Studies need to demonstrate that they meet the eligibility criteria set out in the Department of Health and Social Care policy document “Eligibility for NIHR Clinical Research Network Support (September 2023)”. In order to meet the criteria of open competition and high quality peer review, the funding call from the overseas government must have been open to all qualified individuals in England to apply (as lead or co-applicant), or, where the study is an international collaboration led from overseas, for all qualified individuals within the lead country to apply and for participation to have been open to all qualified sites in England. Such studies have a medium priority for Clinical Research Network support.
E12. My study is funded by a non-commercial (non profit making / charitable) organisation operating solely outside England. Is it eligible for NIHR Clinical Research Network support?
Studies that are funded by a charity operating solely outside of England are considered for eligibility for CRN support via the Non-commercial extended review process. Studies need to demonstrate that they meet the eligibility criteria set out in the Department of Health and Social Care policy document “Eligibility for NIHR Clinical Research Network Support (September 2023)”. In order to meet the criteria of open competition and high quality peer review, the funding call from the Non-commercial organisation must have been open to all qualified individuals in England to apply (as lead or co-applicant), or, where the study is an international collaboration led from overseas, for all qualified individuals within the lead country to apply and for participation to have been open to all qualified sites in England.
E13. My study is funded by an award (fellowship / studentship) from an overseas government/charity. Is this eligible for NIHR Clinical Research Network support?
Research funded following open international competition and high quality peer review by overseas governments or charities is considered for Clinical Research Network (CRN) support via the non-commercial extended review process. In order to meet the criteria of open international competition, the funding call must have been open to all qualified individuals in England to apply (as lead or co-applicant), or, where the study is an international collaboration led from overseas, for all qualified individuals within the lead country to apply and for participation to have been open to all qualified sites in England. Therefore, where a foreign government has funded one of its nationals (ie award made to one individual) to conduct research or training overseas without any element of international competition or collaboration, the study funded by this award will not be eligible for CRN support.
E14. Why is my study being assessed via the extended review process?
The requirement for a study to be assessed through the extended review process is determined by the source of research funding together with the sponsorship arrangements which are in place.
Non-commercial studies (usually sponsored by a University or NHS Trust) which are funded by potentially eligible funding streams undergo additional eligibility checks to ensure the study meets the Department of Health and Social Care policy document “Eligibility for NIHR Clinical Research Network Support (September 2023)”. This is referred to as the non-commercial extended review process.
The following types of non-commercial studies are considered potentially eligible:
- Investigator-initiated, commercial-collaborative studies (Industry-funded, non-industry sponsored studies)
- Non-commercial studies funded by overseas governments
- Non-commercial studies funded by overseas charities
- Certain other high quality studies
As well as ensuring that the study meets the definition of research and full funding for research costs are in place, studies going through the non-commercial extended review process are reviewed by subject specific experts, our NIHR Specialty Leads, against specific criteria focused on:
- Clear value to the NHS, social care or public health
- Taking account of priorities, needs and realities of the NHS, social care or public health.
Our team within the CRN also review against specific criteria focused on:
- Quality (as evidenced by peer review).
- Funding being open to all qualified researchers in England.
Provided that the study meets all of the above requirements then it will be deemed eligible for NIHR CRN Support.
E15. My study is funded and supported by an NIHR Biomedical Research Centre (BRC). Is it eligible for NIHR Clinical Research Network support?
Funding for NIHR Biomedical Research Centres (BRCs) is “self-contained” i.e. funding for both research costs and NHS infrastructure for research (including NHS Support costs) are included in the award. The funding goes directly to the contracted NHS/University partnership and formal partners in each of these NHS/University collaborations. Studies which are fully funded as part of a BRC programme, and take place within the contracted NHS/University partnership and formal partners, will therefore not require additional research infrastructure support from the NIHR CRN. Multi-centre, non-commercial, BRC studies may require NIHR Clinical Research Network support if an additional collaborating site/s (i.e. not the contracted NHS/ University partnership and formal partners) is involved and requires support. In addition, where studies conducted in/led by NIHR BRCs are in receipt of funding from other NIHR research programmes, NIHR Non-commercial Partners or other areas of central government (including research councils), support, including NHS Support Costs, may be sought from the NIHR CRN. Please note that this does not extend to single centre investigator-initiated or industry-collaborative research, and research funded by overseas organisations or ineligible funding streams. The NHS Support Costs for these should be met through the NIHR BRC award (as outlined in the BRC award documentation produced by NIHR). Furthermore, where early translational (experimental medicine) research funded through the BRC funding scheme is conducted within a Clinical Research Facility (CRF) funded by NIHR, the NHS Support Costs associated with the research should be funded from the NIHR BRC funding award. NIHR BRC led studies deemed eligible for ‘additional’ NIHR support from the CRN will be added to the NIHR CRN Portfolio. Recruitment data should be provided for all UK sites (i.e. both the CRN and the contracted NHS/ University partnership and formal partners- BRC sites) and mapped in line with guidance. It is recognised that the requirement for NIHR CRN support may change during the lifecycle of the study, for example a fully funded BRC study (within the contracted NHS/ University partnership and formal partners) may need to open in new CRN supported sites to achieve the study’s recruitment target. Should this situation arise an application for NIHR CRN support should be made following discussion with the local CRN.
E16. My study is funded and supported by the NIHR BioResource, the NIHR Health Informatics Collaborative (HIC) or a Translational Research Collaborations (TRCs). Is it eligible for NIHR Clinical Research Network support?
Additional NIHR Infrastructure support is provided through the NIHR BioResource, NIHR Health Informatics Collaborative (HIC) and eight Translational Research Collaborations (TRCs). These awards are made, via BRC contract variations, to organisations/groups of organisations already in receipt of BRC awards. Therefore the same principles apply to BioResource, HIC and TRC supported studies as to those supported by BRC awards (see E15).
E17. My study is funded and supported by an NIHR Applied Research Collaboration (ARC). Is it eligible for NIHR Clinical Research Network support?
Funding for NIHR Applied Research Collaborations (ARCs) is “self-contained” i.e. funding for both research costs and NHS infrastructure for research (including NHS Support costs) are included in the award. The funding goes directly to the contracted NHS/University partnership and formal partners in each of these NHS/University collaborations. Studies which are fully funded as part of an ARC programme, and within the contracted NHS/University partnership and formal partners, will not therefore require additional research infrastructure support from the NIHR CRN.
Multi-centre, non-commercial, ARC studies, (including ARC-ARC collaborations) may require NIHR Clinical Research Network support if an additional collaborating site/s (i.e. not the contracted NHS/ University partnership and formal partners) is involved and requires support.
In addition, where studies conducted in/led by NIHR ARCs are in receipt of funding from other NIHR research programmes, NIHR Non-commercial Partners or other areas of central government (including research councils), support, including NHS Support Costs, may be sought from the NIHR CRN. Please note that this does not extend to single centre investigator-initiated or industry-collaborative research, and research funded by overseas organisations or ineligible funding streams. The NHS Support Costs for these should be met through the NIHR ARC award.
NIHR ARC led studies deemed eligible for ‘additional’ NIHR support from the CRN will be added to the NIHR CRN Portfolio. Recruitment data should be provided for all UK sites (i.e. both the CRN and the contracted NHS/ University partnership and formal partners - ARC sites) and mapped in line with guidance.
It is recognised that the requirement for NIHR CRN support may change during the lifecycle of the study, for example a fully funded ARC study (within the contracted NHS/ University partnership and formal partners) may need to open in new CRN supported sites to achieve the study’s recruitment target. Should this situation arise an application for NIHR CRN support should be made following discussion with the local CRN.
E18. My study is funded and supported by an NIHR Patient Safety Research Collaborations (PSRCs). Is it eligible for NIHR Clinical Research Network support?
Funding for NIHR Patient Safety Research Centres (PSRCs) is “self-contained” i.e. funding for both research costs and NHS infrastructure for research (including NHS Support costs) are included in the award. The funding goes directly to the contracted NHS/University partnership and formal partners in each of these NHS/University collaborations. Studies which are fully funded as part of a PSRCs programme, and within the contracted NHS/University partnership and formal partners, will not therefore require additional research infrastructure support from the NIHR CRN. Multi-centre, non-commercial, PSRCs studies may require NIHR Clinical Research Network support if an additional collaborating site/s (i.e. not the contracted NHS/ University partnership and formal partners) is involved and requires support. In addition, where studies conducted in/led by NIHR PSRCs are in receipt of funding from other NIHR research programmes, NIHR Non-commercial Partners or other areas of central government (including research councils), support, including NHS Support Costs, may be sought from the NIHR CRN. Please note that this does not extend to single centre investigated-initiated or industry-collaborative research, and research funded by overseas organisations or ineligible funding streams. The NHS Support Costs for these should be met through the NIHR PSRCs award (as outlined in the PSRCs award documentation produced by NIHR). NIHR PSRCs led studies deemed eligible for ‘additional’ NIHR support from the CRN will be added to the NIHR CRN Portfolio. Recruitment data should be provided for all UK sites (i.e. both the CRN and the contracted NHS/ University partnership and formal partners- PSRCs sites) and mapped in line with guidance. It is recognised that the requirement for NIHR CRN support may change during the lifecycle of the study, for example a fully funded PSRCs study (within the contracted NHS/ University partnership and formal partners) may need to open in new CRN supported sites to achieve the study’s recruitment target. Should this situation arise an application for NIHR CRN support should be made following discussion with the local CRN.
E19. My study is funded and supported by an NIHR Medtech and In vitro diagnostic Co-operatives (MICs). Is it eligible for NIHR Clinical Research Network support?
Funding for Medtech and In vitro diagnostic Co-operatives (MICs) is “self-contained” i.e. funding for both research costs and NHS infrastructure for research (including NHS Support costs) are included in the award. The funding goes directly to the contracted NHS/University partnership and formal partners in each of these NHS/University collaborations. Studies which are fully funded as part of a MIC programme, and within the contracted NHS/University partnership and formal partners, will not therefore require additional research infrastructure support from the NIHR CRN.
Multi-centre, non-commercial, MIC studies may require NIHR Clinical Research Network support if an additional collaborating site/s (i.e. not the contracted NHS/ University partnership and formal partners) is involved and requires support.
In addition, where studies conducted in/led by NIHR MICs are in receipt of funding from other NIHR research programmes, NIHR Non-commercial Partners or other areas of central government (including research councils), support, including NHS Support Costs, may be sought from the NIHR CRN. Please note that this does not extend to single centre investigator-initiated or industry-collaborative research, and research funded by overseas organisations or ineligible funding streams. The NHS Support Costs for these should be met through the NIHR MIC award (as outlined in the MIC award documentation produced by NIHR).
NIHR MIC led studies deemed eligible for ‘additional’ NIHR support from the CRN will be added to the NIHR CRN Portfolio. Recruitment data should be provided for all UK sites (i.e. both the CRN and the contracted NHS/ University partnership and formal partners- MIC sites) and mapped in line with guidance.
It is recognised that the requirement for NIHR CRN support may change during the lifecycle of the study, for example a fully funded MIC study(within the contracted NHS/ University partnership and formal partners) may need to open in new CRN supported sites to achieve the study’s recruitment target. Should this situation arise an application for NIHR CRN support should be made following discussion with the local CRN.
E20. My study is supported by another NIHR Infrastructure award but requires additional CRN support, how can I apply for this?
Where possible an application should be made, prior to the study opening
- via the Integrated Research Application System (IRAS). Select "No" to question 5a on the IRAS Project Filter form, this generates question 5b. Select "Yes" to Question 5b “Do you wish to make an application for the study to be considered for NIHR Clinical Research Network (CRN) Support and inclusion in the NIHR Clinical Research Network Portfolio?".
- or via the Non-commercial Portfolio Application service in CPMS.
Visit our CRN Porfolio webpage for more information.
NIHR Infrastructure led studies deemed eligible for ‘additional’ NIHR support from the CRN will be added to the NIHR CRN Portfolio. Recruitment data should be provided for all UK sites (i.e. both the CRN and the contracted NHS/ University partnership and formal partners- Infrastructure sites) and mapped in line with guidance.
It is recognised that the requirement for NIHR CRN support may change during the lifecycle of the study, for example a fully funded NIHR Infrastructure study (within the contracted NHS/ University partnership and formal partners) may need to open in new CRN supported sites to achieve the study’s recruitment target. Should this situation arise an application for NIHR CRN support should be made following discussion with the local CRN.
E21.My study is being conducted within a Clinical Research Facility for Experimental Medicine. Is it eligible for NIHR Clinical Research Network support?
NIHR funding for Clinical Research Facilities (CRFs) does not include funding for research costs. All NIHR CRF supported studies therefore require external funding for research costs which will determine eligibility for NIHR Clinical Research Network Support and inclusion in the NIHR CRN Portfolio.
The purpose of NIHR funding for CRFs is to meet the necessary recurrent NHS infrastructure costs of CRFs. This includes funding to meet NHS Support Costs. Therefore the need for any “additional” NHS support from the Clinical Research Network should be considered on a case-by-case basis at a local level.
To note: where early translational (experimental medicine) research funded through the BRC funding scheme is conducted within a CRF funded by NIHR, the NHS Support Costs associated with the research should be funded from the NIHR BRC funding award.
NIHR CRF supported studies deemed eligible for ‘additional’ NIHR support from the CRN will be added to the NIHR CRN Portfolio. Recruitment data should be provided for all UK sites (i.e. both the CRN and the contracted NHS/ University partnership NIHR CRF sites) and mapped in line with guidance.
It is recognised that the requirement for NIHR CRN support may change during the lifecycle of the study, for example an NIHR CRF supported study may need to open in new CRN supported sites to achieve the study’s recruitment target. Should this situation arise an application for NIHR CRN support should be made following discussion with the local CRN.
E22. My study is being conducted within an Experimental Cancer Medicine Centre. Is it eligible for NIHR CRN support?
Experimental Cancer Medicine Centres (ECMCs) are funded jointly by Cancer Research UK (CRUK), the NIHR in England, and the Health Departments for Scotland, Wales and Northern Ireland.
In England the CRUK/NIHR ECMCs are joint University/NHS partnerships. The purpose of funding is to meet the costs of research infrastructure (via the CRUK element of funding to the University partner) and the costs of NHS infrastructure for research including NHS Support Costs (via the NIHR element of funding to the NHS partner). Funding is not intended to meet the direct research costs of individual studies.
As CRUK is an NIHR Non-commercial Partner, all studies supported by an ECMC award are technically eligible for consideration for NIHR Clinical Research Network support and hence inclusion in the NIHR CRN Portfolio. However, as NIHR funding for ECMCs includes some funding for NHS Support, “additional” NHS support from the Clinical Research Network will be considered on a case-by-case basis at a local level and studies which are fully funded as part of a ECMC programme, and taking place solely within the contracted NHS/University partnership, will not usually be accepted.
Multi-centre, non-commercial, ECMC studies may require NIHR Clinical Research Network support if an additional collaborating site/s is involved and requires support.
In addition, where studies conducted in/led by ECMCs are in receipt of funding from other NIHR research programmes, NIHR Non-commercial Partners or other areas of central government (including research councils), support, including NHS Support Costs, may be sought from the NIHR CRN. Please note that this does not extend to single centre investigator-initiated or industry-collaborative research, and research funded by overseas organisations or ineligible funding streams. The NHS Support Costs for these should be met through the ECMC award. NIHR ECMC led studies deemed eligible for ‘additional’ NIHR support from the CRN will be added to the NIHR CRN Portfolio. Recruitment data should be provided for all UK sites (i.e. both the CRN and the contracted NHS/ University partnership and formal partners- ECMC sites) and mapped in line with guidance.
It is recognised that the requirement for NIHR CRN support may change during the lifecycle of the study, for example a fully funded ECMC study (within the contracted NHS/ University partnership and formal partners) may need to open in new CRN supported sites to achieve the study’s recruitment target. Should this situation arise an application for NIHR CRN support should be made following discussion with the local CRN.
E23. My study is public health research, can it be considered for NIHR Clinical Research Network support?
Yes, the purpose of the NIHR Clinical Research Network (CRN) is to provide infrastructure support for the initiation and delivery of high quality research which benefits the NHS, including relevant research into public health. Public health research studies, regardless of study setting (i.e. conducted within or outside NHS settings), which meet the CRN Eligibility Criteria, are able to receive support through the Clinical Research Network’s Study Support Service. NHS Support (or the equivalent of NHS Support in non-NHS settings) can be provided for attributed activities, as defined by ‘AcoRD’ - the Department of Health and Social Care guidance for attributing the costs of health and social care research. For studies funded by NIHR non-commercial Partner charities who are also members of the Association of Medical Research Charities, support will extend to the provision of ‘Part B Research costs’, as outlined in the AcoRD Frequently Asked Questions.
To be considered eligible, a public health research study must meet the definition of research; have appropriate ethical approval (e.g. NHS Research Ethics Committee (REC), Social Care REC, Ministry of Defence REC, or REC approval from a University); and Health Research Authority (HRA) Approval where required, and have full research funding (i.e. funding to meet all Research Costs in compliance with the AcoRD).
E24. My study is a social care research study taking place in a setting outside of the NHS, is it eligible for NIHR Clinical Research Network support?
Yes, the purpose of the NIHR Clinical Research Network (CRN) is to provide infrastructure support for the initiation and delivery of high quality research which benefits the NHS, including relevant research into social care.
Social care research studies, regardless of study setting (i.e. conducted within or outside NHS settings) are able to receive support through the Clinical Research Network's Study Support Service if they meet CRN Eligibility Criteria.
NHS Support (or the equivalent of NHS Support in non-NHS settings) can be provided for attributed activities, as defined by ‘AcoRD’ - the Department of Health and Social Care guidance for attributing the costs of health and social care research. If your study is funded by a NIHR Non-commercial Partner charity who is also a member of the Association of Medical Research Charities, support will extend to the provision of ‘Part B Research costs’, as outlined in the AcoRD Frequently Asked Questions.
To be considered eligible, your social care research study must meet the definition of research; have appropriate ethical approval (e.g. NHS Research Ethics Committee (REC), Social Care REC, Ministry of Defence REC, or REC approval from a University); and Health Research Authority (HRA) Approval where required, and have full research funding (i.e. funding to meet all Research Costs in compliance with the AcoRD)
E25. How do I apply for NIHR Clinical Research Network support if my study is taking place outside of the NHS and does not require HRA Approval?
Before making an application, you must contact your local CRN who will advise you on what support the CRN can offer for your study. Following this contact you can submit an application through the new Non-commercial Portfolio Application service in CPMS. This service allows investigators to apply earlier and receive an eligibility decision sooner to benefit from the full range of support that our Study Support Service offers.
In order to use the Non-commercial Portfolio Application service, you must first create an account in CPMS. Find out how to create an account and log in to CPMS here .
Before starting your application, it is expected that you will have:
- Discussed your study with your local CRN, through our Study Support Service .
- Confirmed the sponsorship arrangements for your study
- Obtained an IRAS ID for your study by following these steps
- Secured full research funding for your study, in line with the Department of Health and Social Care’s AcoRD policy
You should submit your application as soon as you have secured full research funding for your study, in line with the Department of Health and Social Care’s (DHSC) Attributing the costs of health and social care research (AcoRD) policy. There is no need to wait until you are ready to apply for ethical approvals.
To complete your application you will need to provide a copy of your study protocol and evidence of the research funding you have secured so have these ready before starting your application.
Once we receive your application it will be reviewed against the Department of Health and Social Care’s Eligibility Criteria , and you will be notified of the outcome via email.
E26. My study is already open to recruitment, is it eligible for NIHR Clinical Research Network support?
Although we strongly encourage you to submit your application as soon as you have secured funding, we will accept applications for studies that are at a later stage, e.g. when regulatory approval has been received, or the study has opened to recruitment. Studies in their last six months of recruitment (or, for shorter studies, have less than half the recruitment time left) will not be considered. This is to ensure there is enough time left for the CRN to provide benefit to the delivery of the story.
If your study is open to recruitment, you can submit an application for NIHR Clinical Research Network Support via the Non-commercial Portfolio Application service in CPMS.
In order to use the Non-commercial Portfolio Application service, you must first create an account in CPMS. Find out how to create an account and log in to CPMS on our website .
To complete your application you will need to provide a copy of your study protocol and evidence of the research funding you have secured. Have these ready before starting your application.
Once we receive your application it will be reviewed against the Department of Health and Social Care's Eligibility Criteria and you will be notified of the outcome via email.
E27. My study does not consent participants or the activity does not meet the NIHR CRN definition of recruitment, is it eligible for NIHR CRN support?
Yes. All studies which meet the Eligibility Criteria for Clinical Research Network (CRN) Support are included on the NIHR CRN Portfolio regardless of whether or not the study consents participants or whether the activity meets the NIHR CRN definition of recruitment.
Definition of recruitment
“Recruitment is the enrolment of an individual person meeting specific inclusion criteria into a research study. Each study participant who has both provided informed consent to join a study and is taking part in the study (i.e. participants who count towards the sample size of the study as set out in the study protocol), should be recorded as a participant in our NIHR Central Portfolio Management System (CPMS).”
Part 1. Overview Information
National Institutes of Health ( NIH )
UG1 Clinical Research Cooperative Agreements - Single Project
- August 15, 2024 - Notice of Informational Webinar for Cancer Prevention Clinical Trials Network (CP-CTNet): RFA-CA-24-024 and RFA-CA-24-025. See Notice NOT-CA-24-095 .
- April 4, 2024 - Overview of Grant Application and Review Changes for Due Dates on or after January 25, 2025. See Notice NOT-OD-24-084 .
- August 31, 2022 - Implementation Changes for Genomic Data Sharing Plans Included with Applications Due on or after January 25, 2023. See Notice NOT-OD-22-198 .
- August 5, 2022 - Implementation Details for the NIH Data Management and Sharing Policy. See Notice NOT-OD-22-189 .
See Section III. 3. Additional Information on Eligibility.
Through this Notice of Funding Opportunity (NOFO), the National Cancer Institute (NCI) proposes and will support the Cancer Prevention Clinical Trials Network (CP-CTNet), for which the goals are as follows:
- Design and conduct of early phase clinical trials to assess the safety, tolerability, and cancer preventive potential of agents and interventions of varying classes, many of which target molecules or processes known to be important during carcinogenesis. These trials include phase 0 (micro-dosing), phase I (dose-finding), and phase II (preliminary efficacy) clinical trials.
- Characterization of the effects of these agents and interventions on their molecular targets, as well as on other biological events associated with cancer development (such as cell proliferation, apoptosis, growth factor expression, oncogene expression, immune response) and correlation of these effects with clinical endpoints.
- Development of further scientific insights into the mechanisms of cancer prevention by the agents examined, including the development of novel potential markers as determinants of response.
CP-CTNet consists of two types of components:
- CP-CTNet Sites (covered by companion NOFO, RFA-CA-24-024 ); and
- One CP-CTNet Data Management, Auditing, and Statistical Center (DMASC, covered by this NOFO).
The CP-CTNet Sites will provide scientific leadership in development and conduct of early phase cancer prevention clinical trials as well as in the management and analysis of the data. The DMASC will support the CP-CTNet Sites and coordinate trans-Network activities with the following specific responsibilities:
(i) centralized data management and data reporting,
(ii) clinical trials auditing
(iii) statistical support, and
(iv) administrative support across CP-CTNet.
30 days prior to the application due date
| Application Due Dates | Review and Award Cycles | ||||
|---|---|---|---|---|---|
| New | Renewal / Resubmission / Revision (as allowed) | AIDS - New/Renewal/Resubmission/Revision, as allowed | Scientific Merit Review | Advisory Council Review | Earliest Start Date |
| October 31, 2024 | Not Applicable | Not Applicable | March 2025 | May 2025 | July 2025 |
All applications are due by 5:00 PM local time of applicant organization.
Applicants are encouraged to apply early to allow adequate time to make any corrections to errors found in the application during the submission process by the due date.
No late applications will be accepted for this Notice of Funding Opportunity (NOFO).
Not Applicable
It is critical that applicants follow the instructions in the Research (R) Instructions in the How to Apply - Application Guide , except where instructed to do otherwise (in this NOFO or in a Notice from NIH Guide for Grants and Contracts ).
Conformance to all requirements (both in the How to Apply - Application Guide and the NOFO) is required and strictly enforced. Applicants must read and follow all application instructions in the How to Apply - Application Guide as well as any program-specific instructions noted in Section IV. When the program-specific instructions deviate from those in the How to Apply - Application Guide , follow the program-specific instructions.
Applications that do not comply with these instructions may be delayed or not accepted for review.
There are several options available to submit your application through Grants.gov to NIH and Department of Health and Human Services partners. You must use one of these submission options to access the application forms for this opportunity.
- Use the NIH ASSIST system to prepare, submit and track your application online.
- Use an institutional system-to-system (S2S) solution to prepare and submit your application to Grants.gov and eRA Commons to track your application. Check with your institutional officials regarding availability.
- Use Grants.gov Workspace to prepare and submit your application and eRA Commons to track your application.
Part 2. Full Text of Announcement
Section i. notice of funding opportunity description.
This Notice of Funding Opportunity (NOFO) is intended to fund a Data Management, Auditing, and Statistical Center (DMASC) under the National Cancer Institute (NCI)-supported Cancer Prevention Clinical Trials Network (CP-CTNet). The purpose of CP-CTNet is to perform early phase clinical trials to evaluate the biologic effects of preventive agents and interventions and to determine clinically-relevant correlates in order to advance their development for cancer prevention.
CP-CTNet will support the following two types of components that will be individually awarded through the respective NOFOs indicated below:
- CP-CTNet Sites (covered by companion NOFO, RFA-CA-24-024 ), whose role will be to design, perform and report the results of early phase (phase 0-2) cancer prevention clinical trials.
- Data Management, Auditing, and Statistical Center (DMASC), which will provide centralized data management, statistical support, and auditing of clinical trials data, as well as administrative support for all CP-CTNet recipients ( this NOFO ).
The CP-CTNet DMASC will support the CP-CTNet Sites and coordinate trans-Network activities with the following specific responsibilities:
(i) Centralized data management and data reporting, (ii) Clinical trials auditing, (iii) Statistical support, (iv) Administrative and logistical coordination across CP-CTNet.
To achieve these goals, the DMASC is expected to provide multi-disciplinary expertise in information technology, clinical research informatics, clinical trials auditing, clinical trials methodology and biostatistics, and operations management to support CP-CTNet activities.
Key Definitions for the context of this NOFO:
- The terms “Clinical Research” and “Clinical Trials” in this NOFO follow the NIH definitions ( https://grants.nih.gov/policy/clinical-trials/glossary-ct.htm#ClinicalResearch .)
- CP-CTNet Site: UG1 Consortium supported by RFA-CA-24-024 consisting of the Lead Academic Organization (LAO, recipient of the UG1 award) and Affiliated Organizations (AOs). Each LAO serves as the research hub for the CP-CTNet Site. A LAO may have parts that represent the same legal entity, even if their location is different from the main location of the awardee (such parts are referred to as "LAO integrated components"). AOs are any institutions collaborating with the LAO on clinical trials under sub-contractual/consortium arrangements.
- Study Chair: the investigator who leads a given clinical trial
- Target organ: organ of focus for a given clinical trial. The clinical trial will provide information about prevention or interception of cancer arising from the specific target organ.
- NCI Central Institutional Review Board (CIRB): a centralized approach to human subject protection through a process that streamlines local IRB review of selected NCI-sponsored trials for institutions across the country by relying on national experts to ensure trials are reviewed efficiently and with the highest ethical and quality standards ( https://www.ncicirb.org/about-cirb/ ).
- An Accrued Participant is defined as an individual who has completed the informed consent process, has been deemed eligible through all levels of the screening process, and has started the study intervention (e.g., actually received the agent and/or intervention to be tested).
- A Screened Participant is defined as an individual who has signed consent to proceed with evaluation for eligibility for a study after preliminary eligibility (e.g., presence of a risk factor for a specific cancer, history of a specific premalignant lesion, etc.) has been determined.
- Cross-network trial: a trial performed across two or more CP-CTNet Sites such that funding for the trial is provided from the grants to all of the participating CP-CTNet Sites.
Note: It is essential that applicants for this NOFO are also fully familiar with the companion NOFO, RFA-CA-24-024 , including the specific goals and other vital details of their functioning within the CP-CTNet.
The search for effective cancer preventive agents, in the context of a rapidly advancing molecular understanding of the process of carcinogenesis, has led to the study of an increasing number of agents that intervene in specific molecular pathways thought to be critical to cancer development. The rapidly advancing, albeit incomplete, understanding of the early phases of cancer development provides a strong rationale for increased investment in cancer prevention. There has been a longstanding interest in infectious etiologies of cancer and highly effective preventive vaccines (human papillomavirus, hepatitis B) or treatments (hepatitis C) are now integrated into standard care. Targeting these obligatory causes of cancer has profound consequences on public health. For instance, in the case of human papillomavirus, an effective country-wide vaccination program in the UK led to a highly significant population-level decrease in cervical cancer, as high as 97% when vaccinations were given at age 12-13 years. More recently, the recognition of the importance of the role of the immune system in tumor development and the recent successes in cancer immunotherapy for the treatment of advanced malignancies have led to a resurgence of interest in immunopreventive approaches aimed at cancers not associated with infections. The increasing number and molecularly or immunologically targeted nature of new agents require an efficient clinical trials system for evaluation and screening prior to moving to larger definitive phase III trials. These complex trials must also include extensive biomarker analyses, investigation of the biologic effects of the agent on the intended target, and correlation with clinically relevant indicators of potential health outcomes.
The nature of cancer prevention clinical trials requires access to specialized high-risk populations who obtain their care from different subspecialists and expertise in tissue collection and biomarker analysis. A typical phase II trial might examine the effect of an intervention on a histologically-proven premalignancy in participants at risk for cancer. This requires the screening of multiple high-risk individuals with invasive procedures, such as a colonoscopy or bronchoscopy, to identify those who harbor such premalignancies, followed by post-treatment procedures with biopsies to assess the intervention’s efficacy. Other types of studies employed in cancer preventive agent development include (but are not limited to): phase 0 micro-dosing trials, phase I pharmacokinetic and pharmacodynamic trials, and window-of-opportunity trials performed prior to definitive cancer treatment. Individuals participating in such studies include healthy volunteers, individuals at high risk for cancer either due to genetic predisposition or the presence of premalignant lesions, and cancer patients either prior to or after definitive treatment of the primary malignancy. Thus, multi-institutional groups of clinicians from diverse specialties, research nurses, pathologists, translational scientists, statisticians, data managers, and other personnel with expertise in cancer prevention, drug development, and biomarker analysis are needed to successfully perform increasingly complex cancer prevention clinical trials.
NCI supports the systematic development of cancer preventive agents through three major programmatic initiatives managed by the Division of Cancer Prevention (DCP):
- Preclinical agent development is supported through the PREVENT Cancer Preclinical Drug Development Program and the Cancer Prevention-Interception Targeted Agent Discovery Program ( CAP-IT ).
- The early phase clinical development (phase 0-II clinical trials) of promising cancer preventive agents is supported by the Cancer Prevention Clinical Trials Network ( CP-CTNet ). The goal of this network is to identify preliminary efficacy and safety of preventive strategies. The results of preliminary efficacy studies provide important data to inform decisions to proceed to lengthy, expensive phase III cancer prevention trials, thereby minimizing the risk of failure in phase III. This program fills a critical niche in cancer preventive drug development that is poorly supported by the pharmaceutical industry and not well represented in the investigator-initiated grants portfolio.
- The phase III cancer preventive agent development, aimed at demonstrating definitive efficacy, is supported via the NCI Community Oncology Research Program (NCORP; https://ncorp.cancer.gov/ ).
This NOFO and RFA-CA-24-024 (for CP-CTNet Sites) will fund the continuation of CP-CTNet. The proposed DMASC (this NOFO) will replace and expand activities performed previously by the CP-CTNet Data Management, Auditing, and Coordinating Center (DMACC) (U24) (funded previously under RFA-CA-18-030 ).
Overall Goals of CP-CTNet
The overall goal of CP-CTNet is to perform early phase cancer prevention clinical trials to identify agents and interventions that can advance through the various phases of clinical development. Specific objectives are summarized below:
- To efficiently design and conduct early phase clinical trials to assess the cancer preventive potential of a variety of different agents or strategies. Emphasis is on novel agents and interventions that target relevant pathways important in carcinogenesis.
- To characterize the effects of these agents and interventions on their molecular targets, immune function, and other biological events associated with cancer development (e.g., cell proliferation, apoptosis, growth factor expression, oncogene expression, etc.) and correlate these effects with clinical endpoints.
- To develop further scientific insights into the mechanism of cancer prevention by the agent or strategy examined and to continue to develop novel potential markers as determinants of response.
- To facilitate development and conduct of cross-network trials and to speed up preventive agent development.
Roles of CP-CTNet components :
Each CP-CTNet Site will perform a variety of early phase cancer prevention trials in appropriately high-risk populations, ranging from phase 0 to phase IIb trials. Agents to be studied will include those developed by the pharmaceutical industry and provided to NCI for collaborative development, commercially available agents, agents developed by the grantees, and agents developed by NCI.
Each CP-CTNet LAO will be expected to serve as the main research infrastructure to support the performance of clinical trials. The LAOs will provide administrative support and oversight to clinical trial performance across their member AOs, as well as develop and perform specific clinical trials within their own (LAO) institutions. The role of the AOs will be to develop clinical trials in collaboration with the LAOs and to accrue to specific studies. LAOs and AOs may participate in studies arising within their CP-CTNet Site as well as within other CP-CTNet Sites. Studies that are performed across CP-CTNet Sites, with funding provided by different CP-CTNet Sites, are considered cross-network trials and are encouraged.
The LAO will be required to interact closely with the DMASC, which will be responsible for support for clinical trials data management and reporting, clinical trials auditing, statistical support, and administrative and logistical coordination across CP-CTNet, as described below.
Because of the DMASC role in the Network, it is essential that the DMASC applicants are fully familiar not only with this NOFO, but also with the companion NOFO, RFA-CA-24-024 , including the specific goals and requirements for the CP-CTNet Sites and other vital details of their functioning within the CP-CTNet.
DMASC Goals and Scope of Activities
Functional Areas.
The DMASC will be expected to facilitate efficient conduct of CP-CTNet clinical trials by providing support in four key functional areas:
- Centralized data management and data reporting;
- Clinical trials auditing;
- Statistical support, and
- Administrative and logistical coordination across CP-CTNet.
To achieve these goals, the DMASC should have appropriate multi-disciplinary expertise, background, skills, as well as established infrastructure for all the applicable areas of activities:
- Information technology;
- Clinical research informatics;
- Clinical trials auditing;
- Clinical trials methodology and biostatistics; and
- General administrative and logistical coordination capabilities to support CP-CTNet activities.
The main DMASC responsibilities in each of four key functional areas defined above include, but are not limited, to the following aspects:
Centralized Data Management and Data Reporting:
- Providing centralized data management for CP-CTNet clinical trials using Medidata Rave® as the NCI-designated Clinical Data Management System (CDMS) of record. The applicant is expected to create and enforce data management policies, formulate management techniques for quality data collection to ensure adequacy, integrity, and legitimacy of data, and devise and implement secure procedures for data management and analysis with attention to all technical and regulatory aspects.
- Providing data management support for tracking and improving participant accrual in CP-CTNet clinical trials by developing and/or managing tools and resources for the collection, review, analysis, reporting, and quality improvement for accrual data and related information in partnership with CP-CTNet Sites and NCI staff.
- Supporting routine and ad-hoc reporting of clinical trials data to CP-CTNet Sites and to the NCI. The awardee will be expected to develop reports using pre-designed and custom formats that utilize real-time, historical, auditing, and/or analytical information.
- Developing web-services (e.g., Representational State Transfer (REST), application programming interface (API), etc.) for system-to-system data exchange. The awardee will be expected to develop web-services using industry best practices to share clinical trial data with CP-CTNet Sites and with the NCI.
- Supporting efforts in management, tracking, and storage of biospecimens from CP-CTNet trials by maintaining a virtual biospecimen data inventory system that interfaces with CP-CTNet Sites and the DCP Biorepository Program. The awardee team will be expected to manage a centralized electronic inventory that will permit real-time remote access to status of biospecimens from CP-CTNet clinical trials.
Clinical Trials Auditing:
- Conducting independent auditing of clinical trials data and processes at all CP-CTNet LAOs and AOs to ensure that all relevant good clinical practice (GCP) guidelines, protocol requirements, applicable regulatory requirements, federal regulations, and NIH/NCI/DCP policies are followed.
- Interacting with CP-CTNet Sites and NCI staff to identify areas for systemic and policy-level improvements in order to increase both efficiency and compliance, ensure the protection of human subjects, and enhance the quality and integrity of CP-CTNet clinical trials.
Statistical Support
- Providing statistical consultation and expertise for all CP-CTNet trials
- Serving as primary statisticians of record for all cross-network clinical trials and providing support to CP-CTNet LAOs and AOs
Administrative and Logistical Coordination:
- The DMASC will provide support for administrative and logistical coordination across CP-CTNet operations. The applicant will establish and maintain a unified and coordinated operational structure, including processes and documentation that appropriately support and integrate the logistical and administrative requirements of CP-CTNet.
- The DMASC will provide support for the development, presentation, and dissemination of educational materials and other capacity building resources for CP-CTNet recruitment and retention activities.
Required Functional Components: To realize the stated objectives, the CP-CTNet DMASC applicants must plan for organizing the following four required functional components:
- Data Management and Reporting Unit – This functional component must encompass experienced personnel providing support and leadership for activities in data management, data reporting, and web-services via management and coordination with teams of information technology and healthcare professionals at all levels. The emphasis is a team approach for efficient data management and reporting/data exchange functions in support of CP-CTNet clinical trials.
- Clinical Trials Auditing Unit – This functional component must encompass activities and personnel to conduct remote/virtual and on-site auditing of clinical trials data and processes for all CP-CTNet LAOs and AOs, in compliance with good clinical practice and applicable regulatory requirements, federal regulations, and NIH, NCI and DCP policies.
- Statistical Unit– This functional component must encompass activities and personnel to provide statistical expertise and consultation for all CP-CTNet trials and serve as the primary statisticians of record for all cross-network clinical trials in support of CP-CTNet LAOs and AOs.
- Administrative and Logistical Coordinating Unit - This functional component must encompass activities that establish and maintain a unified and coordinated operational structure, including processes and documentation to appropriately support and integrate the logistical, administrative, and resource development requirements of CP-CTNet.
CP-CTNet Network Governance and Trans-Network Activities
Steering Committee: The representatives of CP-CTNet awardees (with the participation of the NCI) will be expected to form a Steering Committee as a self-governing body for the Network. For details on the composition and responsibilities of the Steering Committee, see Section VI.2 under Cooperative Agreement Terms and Conditions of the Award.
Trans-Network Research Activities
The DMASC and CP-CTNet Sites will be expected to work jointly toward the CP-CTNet network goals by:
- Collaborating on network activities;
- Working with network collaborators to develop cross-network clinical trials; and
- Participating in high priority ancillary studies, including developmental work for new biomarkers.
Non-responsive Applications
The following types of activities remain outside the scope of this NOFO, and applications proposing them are non-responsive to this NOFO and will not be reviewed:
- Proposals that do not cover each of the four functional areas of DMASC activities, viz., data management and reporting, clinical trials auditing, statistical support, and administrative and logistical coordination.
Additional NCI Support for the Initiative (beyond the scope of the two CP-CTNet NOFOs).
NCI/DCP will provide the following additional infrastructure support to CP-CTNet participants:
- Regulatory support, including Investigational New Drug (IND) application preparation and Food and Drug Administration (FDA) reporting
- Agent acquisition, packaging, and distribution, including placebo manufacture when feasible
- Central Institutional Review Board (CIRB) review
- Protocol receipt, review, and approval process, and study document submissions and management
NCI will support data and specimen access through the Cancer Data Access System ( CDAS ). This portal will provide access to the CP-CTNet clinical trial data after trial publication and will provide access to request associated residual biospecimens. NCI will provide long-term storage for the collected biospecimens.
See Section VIII. Other Information for award authorities and regulations.
Investigators proposing NIH-defined clinical trials may refer to the Research Methods Resources website for information about developing statistical methods and study designs.
Section II. Award Information
Cooperative Agreement: A financial assistance mechanism used when there will be substantial Federal scientific or programmatic involvement. Substantial involvement means that, after award, NIH scientific or program staff will assist, guide, coordinate, or participate in project activities. See Section VI.2 for additional information about the substantial involvement for this NOFO.
The OER Glossary and the How to Apply - Application Guide provides details on these application types. Only those application types listed here are allowed for this NOFO.
Required: Only accepting applications that propose clinical trial(s).
Need help determining whether you are doing a clinical trial?
NCI intends to commit $3,250,000 (total costs) each year for 6 years beginning in FY2025 to support one award for the CP-CTNet DMASC.
The requested budget must not exceed $2,000,000 in direct costs in any single year.
All applicants should request a 6-year project period.
NIH grants policies as described in the NIH Grants Policy Statement will apply to the applications submitted and awards made from this NOFO.
Section III. Eligibility Information
1. eligible applicants eligible organizations all organizations administering an eligible parent award may apply for a supplement under this nofo. higher education institutions public/state controlled institutions of higher education private institutions of higher education the following types of higher education institutions are always encouraged to apply for nih support as public or private institutions of higher education: hispanic-serving institutions historically black colleges and universities (hbcus) tribally controlled colleges and universities (tccus) alaska native and native hawaiian serving institutions asian american native american pacific islander serving institutions (aanapisis) nonprofits other than institutions of higher education nonprofits with 501(c)(3) irs status (other than institutions of higher education) nonprofits without 501(c)(3) irs status (other than institutions of higher education) for-profit organizations small businesses for-profit organizations (other than small businesses) local governments state governments county governments city or township governments special district governments indian/native american tribal governments (federally recognized) indian/native american tribal governments (other than federally recognized) federal government eligible agencies of the federal government u.s. territory or possession other independent school districts public housing authorities/indian housing authorities native american tribal organizations (other than federally recognized tribal governments) faith-based or community-based organizations regional organizations foreign organizations non-domestic (non-u.s.) entities (foreign organizations) are not eligible to apply. non-domestic (non-u.s.) components of u.s. organizations are not eligible to apply. foreign components, as defined in the nih grants policy statement , are allowed. required registrations applicant organizations applicant organizations must complete and maintain the following registrations as described in the how to apply - application guide to be eligible to apply for or receive an award. all registrations must be completed prior to the application being submitted. registration can take 6 weeks or more, so applicants should begin the registration process as soon as possible. failure to complete registrations in advance of a due date is not a valid reason for a late submission, please reference nih grants policy statement section 2.3.9.2 electronically submitted applications for additional information. system for award management (sam) – applicants must complete and maintain an active registration, which requires renewal at least annually . the renewal process may require as much time as the initial registration. sam registration includes the assignment of a commercial and government entity (cage) code for domestic organizations which have not already been assigned a cage code. nato commercial and government entity (ncage) code – foreign organizations must obtain an ncage code (in lieu of a cage code) in order to register in sam. unique entity identifier (uei) - a uei is issued as part of the sam.gov registration process. the same uei must be used for all registrations, as well as on the grant application. era commons - once the unique organization identifier is established, organizations can register with era commons in tandem with completing their grants.gov registration; all registrations must be in place by time of submission. era commons requires organizations to identify at least one signing official (so) and at least one program director/principal investigator (pd/pi) account in order to submit an application. grants.gov – applicants must have an active sam registration in order to complete the grants.gov registration. program directors/principal investigators (pd(s)/pi(s)) all pd(s)/pi(s) must have an era commons account. pd(s)/pi(s) should work with their organizational officials to either create a new account or to affiliate their existing account with the applicant organization in era commons. if the pd/pi is also the organizational signing official, they must have two distinct era commons accounts, one for each role. obtaining an era commons account can take up to 2 weeks. eligible individuals (program director/principal investigator) any individual(s) with the skills, knowledge, and resources necessary to carry out the proposed research as the program director(s)/principal investigator(s) (pd(s)/pi(s)) is invited to work with their organization to develop an application for support. individuals from diverse backgrounds, including underrepresented racial and ethnic groups, individuals with disabilities, and women are always encouraged to apply for nih support. see, reminder: notice of nih's encouragement of applications supporting individuals from underrepresented ethnic and racial groups as well as individuals with disabilities, not-od-22-019 . for institutions/organizations proposing multiple pds/pis, visit the multiple program director/principal investigator policy and submission details in the senior/key person profile (expanded) component of the how to apply - application guide . the individual designated as pd/pi for the cp-ctnet dmasc must have their primary affiliation at the application-submitting institution. for proposals with multiple pds/pis, the second individual designated as mpi may have a primary affiliation at a different us institution. these individuals are expected to be nationally and internationally recognized leaders in management and stewardship of network-based/multicenter clinical trials. their expertise should broadly encompass the four functional areas of dmasc activities, viz., data management and reporting, clinical trials auditing, statistical support, and administrative and logistical coordination. it is expected that the pds/pis lead a multi-disciplinary team comprised of domain-specific experts with substantial experience in leading each of the four functional areas of the cp-ctnet dmasc. the pds/pis of applications submitted in response to this nofo must not be named as senior/key personnel or other significant contributors on any teams submitting applications to the companion nofo, rfa-ca-24-024 . 2. cost sharing.
This NOFO does not require cost sharing as defined in the NIH Grants Policy Statement Section 1.2 Definition of Terms .
3. Additional Information on Eligibility
Number of Applications
Applicant organizations may submit more than one application, provided that each application is scientifically distinct.
The NIH will not accept duplicate or highly overlapping applications under review at the same time, per NIH Grants Policy Statement Section 2.3.7.4 Submission of Resubmission Application . This means that the NIH will not accept:
- A new (A0) application that is submitted before issuance of the summary statement from the review of an overlapping new (A0) or resubmission (A1) application.
- A resubmission (A1) application that is submitted before issuance of the summary statement from the review of the previous new (A0) application.
- An application that has substantial overlap with another application pending appeal of initial peer review (see NIH Grants Policy Statement 2.3.9.4 Similar, Essentially Identical, or Identical Applications ).
Section IV. Application and Submission Information
1. requesting an application package.
The application forms package specific to this opportunity must be accessed through ASSIST, Grants.gov Workspace or an institutional system-to-system solution. Links to apply using ASSIST or Grants.gov Workspace are available in Part 1 of this NOFO. See your administrative office for instructions if you plan to use an institutional system-to-system solution.
2. Content and Form of Application Submission
It is critical that applicants follow the instructions in the Research (R) Instructions in the How to Apply - Application Guide except where instructed in this notice of funding opportunity to do otherwise. Conformance to the requirements in the How to Apply - Application Guide is required and strictly enforced. Applications that are out of compliance with these instructions may be delayed or not accepted for review.
Letter of Intent
Although a letter of intent is not required, is not binding, and does not enter into the review of a subsequent application, the information that it contains allows IC staff to estimate the potential review workload and plan the review.
By the date listed in Part 1. Overview Information , prospective applicants are asked to submit a letter of intent that includes the following information:
- Descriptive title of proposed activity
- Name(s), address(es), and telephone number(s) of the PD(s)/PI(s)
- Names of other key personnel
- Participating institution(s)
- Number and title of this funding opportunity
The letter of intent should be sent to:
Eva Szabo, MD Telephone: 240-276-7011 Fax: 240-276-7848 Email: [email protected]
All page limitations described in the How to Apply – Application Guide and the Table of Page Limits must be followed.
, with the following exceptions or additional requirements:
For this specific NOFO, the Research Strategy section is limited to 30 pages.
The following section supplements the instructions found in the How to Apply – Application Guide and should be used for preparing an application to this NOFO.
All instructions in the How to Apply - Application Guide must be followed.
Other Attachments: Applicants must provide the following additional material specified below. Each attachment should be uploaded as a separate PDF using the indicated filenames (which will serve as application bookmarks).
Attachment 1. Completed and Ongoing Data Management and Reporting Projects (Use filename: Data Management Projects): List all ongoing and completed data management and reporting projects that the applicant team has led and coordinated during the last 5 years. A table can be used to show such information as project title, data management scope, data reporting scope, years project was/is open, and additional columns for significant and relevant project outcomes.
Attachment 2. Completed and Ongoing Clinical Trials Auditing Projects (Use filename: Auditing Projects): List all ongoing clinical trials auditing projects as well as those completed during the last 5 years. Projects conducted by or coordinated by the DMASC applicant institution(s) as well as projects conducted by affiliated organizations should be included. A table can be used to show such information as project title, focus area/organ system and phase of clinical trials, types/frequency/duration of auditing, years project was/is open, and additional columns for significant project outcomes.
Attachment 3. Completed and Ongoing Statistical Support Projects (Use filename: Statistical Projects): List all ongoing statistical support projects as well as those completed during the last 5 years. Projects conducted by or coordinated by the DMASC applicant institution(s) as well as projects conducted by affiliated organizations should be included. A table can be used to show such information as project title, focus area/organ system and phase of clinical trials, nature/scope of statistical support, years project was/is open, and additional columns for significant project outcomes.
Attachment 4. Completed and Ongoing Multi-Site Coordination Projects (Use filename: Coordination projects): List all ongoing network/multi-site trial coordination projects as well as those completed during the last 5 years. Projects conducted by or coordinated by the DMASC applicant institution(s) as well as projects conducted by affiliated organizations should be included. A table can be used to show such information as project title, focus area/organ system and phase of the clinical trial network/multi-site trial, scope of coordination activities, years project was/is open, and additional columns for significant project outcomes.
R&R Budget
Important Note on Budget : The requested budget for the DMASC should not include costs for standardized central operational, regulatory, and administrative support provided by the NCI or existing NCI Contractors (details provided at http://prevention.cancer.gov/cp-ctnet ) or the tasks/activities to be performed by the CP-CTNet Sites (LAOs/AOs) as defined in the companion RFA-CA-24-024 .
Personnel Effort
- PD/PI Effort Commitment. A minimum effort level of 1.8 person-months is required for the designated DMASC contact PD/PI. In case of a multi-PDs/PIs application, each PD/PI will be required to commit a minimum of 1.2 person-months. This level cannot be reduced during the project period.
- DMASC Functional Unit Directors. It is anticipated that individuals serving as DMASC Functional Unit Directors will commit substantial efforts (e.g., minimum 1.2 person-months) to this role.
DMASC Functional Units
- Costs for hosting, maintaining, and providing round-the-clock access for an instance of Medidata Rave as the Clinical Data Management System (CDMS) of record (the license and specifications for the CDMS will be provided by the NCI).
- Costs for the various activities described in the Research Plan including, but not limited to, the development of data management policies, formulating management techniques for quality data collection to ensure adequacy, integrity and legitimacy of data, and devising and implementing secure procedures for supporting clinical trials data management and analysis with attention to all technical and regulatory aspects, developing reports to perform clinical trial oversight, data review and/or program support, and end-of-study data collection and reporting requirements for the final study closeout process.
- Costs for developing web services to exchange data between systems to assist with knowledge transfer and/or portfolio management, and for regulatory reporting.
- Costs for plans to provide data management and reporting support for NCI's efforts in tracking and improving participant accrual in CP-CTNet clinical trials ( DCP’s Accrual Quality Improvement Program, AQuIP ).
- Costs for maintaining a virtual biospecimen inventory system for CP-CTNet that interfaces with CP-CTNet Sites and the DCP Biorepository Program.
- LAOs will be audited on-site annually, and as needed, remotely.
- All AOs will be audited once they have accrued 3 or more participants for a given clinical trial.
- For intermediate and low-risk trials (e.g., FDA approved agent, over the counter medication or supplement, agent with low toxicity profile), follow-up audits will be conducted every 3 years on > 50% of AOs meeting the accrual threshold (?3 participants). Site selection criteria may include but not be limited to protocol and data entry compliance, stable site staff, past audit performance rating, etc.
- For high-risk trials (e.g., first in human trial, trial with moderate toxicity expected and/or post-administration observation period required, etc.), follow-up audits for all AOs will be conducted at least every three years, or more frequently as determined by DCP.
- Expected ‘for-cause’ on-site auditing may be conducted on an ad hoc basis.
- Note: remote audits may be considered in specific instances, with DCP concurrence
- Personnel support and related costs for providing statistical expertise and consultation for all CP-CTNet trials
- Personnel costs for serving as primary statisticians on the record for all cross network clinical trials in support of CP-CTNet LAOs and AOs.
- Personnel support for coordination and standardization of CP-CTNet operational activities.
- Costs for development and maintenance of documents, website content, and other educational and training materials
- Costs for hosting leadership conference calls every two months, ad hoc conference calls, Steering Committee meetings
- Costs for providing logistical and administrative support (e.g. web registration, on-site logistics support, etc.) for the CP-CTNet annual in-person meeting and other meetings as required.
- Travel funds for DMASC personnel to travel to one annual investigator meeting per year in Rockville, MD (see section VI.2)
Restricted Funds for Support of Young/Emerging Investigators
An amount of $125,000 per year (direct costs) should be entered as " Restricted Funds " under the "Other Expenses" category in the Budget form for years 1-5. These funds are intended for mentorship and professional development of young/emerging investigators at the CP-CTNet Sites (e.g., faculty, clinical fellows, post-doctoral fellows), including opportunities to lead clinical trials. Although investigators will be affiliated with CP-CTNet Sites, the funds will be administered by the DMASC, with a budget of $25,000 per year reserved per CP-CTNet Site, with details not known at the time of application for this NOFO. Specific projects to use these funds will be proposed post-award and will be subject to Steering Committee approval.
The following costs should not be included in the budget:
- Costs associated with routine patient care that are reimbursable by insurance.
- Costs for alterations and renovations.
- Costs for activities to be provided by the CP-CTNet Sites or covered by services/functions funded directly by NCI outside of this NOFO (listed in Section I).
Budget Justification Additional Instructions:
- In addition to the standard items for this attachment, provide a breakdown of the direct costs to show separate amounts for each functional unit of the DMASC (i.e., Data Management and Reporting Unit, Clinical Trials Auditing Unit, Statistical Support Unit, Administrative and Logistical Coordinating Unit), and the amount allocated to the Restricted Funds.
R&R Subaward Budget
All instructions in the How to Apply - Application Guide must be followed.
All instructions in the How to Apply - Application Guide must be followed, with the following additional instructions:
Specific Aims : Describe the overall Specific Aims for the proposed CP-CTNet DMASC addressing the applicant's goals for providing support for centralized data management and reporting, clinical trials auditing, statistical support, and coordination of operations and activities across CP-CTNet.
Research Strategy : Organize the overall Research Strategy section into the following sub-sections in the specified order and follow instructions provided below. Start each sub-section with the appropriate sub-section heading.
Subsection A: DMASC Overview and Capabilities
- Provide an overview of the proposed DMASC in the context of its role in the CP-CTNet and its primary responsibilities identified in Section I, indicating how DMASC can facilitate and enhance clinical trials conducted across CP-CTNet.
- Highlight any unique approaches of the proposed DMASC that reflect innovative ways of coordinating multi-institutional, trans-disciplinary, clinical trials research.
- Demonstrate ability to handle and manage multi-institutional clinical trials data, including the use of cancer-relevant common data elements (CDEs), standardized procedures for data collection, data quality control/quality assurance, etc.
- Demonstrate ability to provide data management support for targeted clinical trials enhancement initiatives such as accrual tracking, biospecimen data management, etc.
- Demonstrate ability to provide data reporting and web-services.
- Outline ability to plan, manage, and implement multi-stage clinical trials auditing implementation frameworks.
- Demonstrate ability to provide statistical consultation and expertise and design and implement statistical analysis plans as the lead statistician(s) on cross-network, multi-center clinical trials
- Outline coordination and organizational support for multi-institutional and multi-disciplinary collaborative research efforts
- Describe how the four key required DMASC Functional Units (plans for which will be described in Subsections B-F below) will contribute to aspects, attributes, and functions of CP-CTNet identified in Section I. The description must address (and be consistent with) the requirements of the NCI DCP CP-CTNet Standard Operation Procedures ( https://prevention.cancer.gov/clinical-trials/clinical-trials-management/cp-ctnet-instructions-forms-and-templates ) for the conduct of prevention clinical trials.
Note: Supporting documentation for this sub-section is requested under Other Attachments (Attachments 1-4).
Subsection B: Data Management and Reporting Unit:
Describe proposed approach and plans for efficient management of clinical trials data collected within CP-CTNet. Specifically, address support activities for the following:
1. Management of Clinical Trials Data
Describe plans for creating and enforcing data management policies, formulating management techniques for quality data collection to ensure adequacy, integrity and legitimacy of data, and devising and implementing secure procedures for clinical trials data management and analysis with attention to all technical and regulatory aspects. These should specifically address the following:
- General description for hosting and maintaining an instance of Medidata Rave as the Clinical Data Management System (CDMS) of record. The license and specifications for the CDMS will be provided by the NCI. (Additional information can be found at http://prevention.cancer.gov/cp-ctnet .)
- Procedures to ensure that the system is available 7 days per week with minimum interruptions to reduce the risk of data loss and disruption of operations.
- Procedures to ensure all CDMS systems and modules involving clinical trials data comply with Part 11 of Title 21 of the Code of Federal Regulations: Electronic Records; Electronic Signatures;
- Plans for data management activities including but not limited to CDMS study build tasks to develop and/or maintain standardized and study-specific eCRFs. NCI-defined common data elements (CDEs) and other CDMS elements should be utilized to ensure data consistency, quality and integrity across studies.
- Data standards and clinical data requirements to be defined and established for CP-CTNet clinical trials
- Procedures for the electronic transfer of data that meet or exceed the NIH and NCI data security standards and guidelines.
- Education and training required for the efficient use of Medidata Rave or other databases by CP-CTNet and NCI staff as needed.
- System set-up and integration with other NIH, NCI and DCP systems. (Additional information can be found at http://prevention.cancer.gov/cp-ctnet.)
- Ad hoc support for AQuIP data reports and/or graphs, or other data support for presentations, manuscript preparation, and other publications.
2. Routine and Ad-hoc Reporting of Clinical Trials Data
Outline plans for developing reports to perform clinical trial oversight, data review and/or program support, by addressing the following:
- Development and/or maintenance of standard operating procedures for clinical trials data reporting, including but not limited to transfer of data captured from Medidata Rave or other databases to DCP monthly, bi-annually, and ad-hoc. (Additional information can be found at http://prevention.cancer.gov/cp-ctnet .)
- Provide routine and ad-hoc reports to assess data quality, timeliness and completeness, assist in the resolution of data discrepancies, and present pertinent clinical trial information to CP-CTNet and NCI staff.
- Support of end-of-study data collection and reporting requirements for the final study closeout process. (Additional information can be found at http://prevention.cancer.gov/cp-ctnet .)
3. Development of Web-Services
Outline plans for developing web services to exchange data between systems to assist with knowledge transfer and/or portfolio management, by addressing the following:
- Development of web-services (e.g., Representational State Transfer (REST), application programming interface (API), etc.) for system-to-system data exchange using industry best practices to exchange clinical trial data.
- Development and implementation of web-services for NCI to receive electronic transfer of standardized dataset from Medidata Rave or other databases to assist NCI with complying with NIH and/or other Federal-level data reporting requirements. (Additional information can be found at http://prevention.cancer.gov/cp-ctnet.)
4. Data Management for Accrual Tracking
Describe plans for providing data management support for NCI's efforts in tracking and improving participant accrual in CP-CTNet clinical trials.
- Develop and/or maintain tools, system(s), and processes for the collection, review, analysis, and reporting of per-person accrual and related information for clinical trials in partnership with CP-CTNet performance sites.
- Provide data management and reporting support for NCI's efforts in tracking and improving participant accrual in CP-CTNet clinical trials (via DCP’s Accrual Quality Improvement Program (AQuIP ).
5. Virtual Biospecimen Inventory System for Clinical Trials
Describe plans for supporting NCI's efforts in management, tracking, and storage of biospecimens from CP-CTNet clinical trials.
- Outline plans for developing and maintaining a virtual biospecimen inventory system for CP-CTNet that interfaces with CP-CTNet Sites and the DCP Biorepository Program. The plan should include the approach for a seamless and secure transfer of the virtual biospecimen data/inventory from the current U24 DMACC grantee. (Additional information can be found at http://prevention.cancer.gov/cp-ctnet .).
- The virtual inventory system should include standard clinical and specimen annotations and study-specific information and should have remote (web-based) real-time access functionality.
Subsection C: Clinical Trials Auditing Unit:
Describe plans and approaches for audits of all CP-CTNet Sites that DMASC will be required to perform as an entity outside of the audited sites. The plans should ensure that all clinical trial-related activities CP-CTNet Sites are in compliance with federal regulations, protocol requirements, GCP guidelines, and NIH, NCI and DCP policies, procedures and requirements for protocol and clinical trials development, implementation and management. The plans need to accommodate the following required to audit frequency:
- For intermediate and low-risk trials (e.g., FDA approved agent, over the counter medication or supplement, agent with low toxicity profile), follow-up audits will be conducted every 3 years on > 50% of AOs meeting the accrual threshold (?3 participants). Site selection criteria may include but not be limited to protocol and data entry compliance, stable site staff, past audit performance rating, etc.)
- For high-risk trials (e.g., first in human trial, trial with moderate toxicity expected and/or post-administration observation period required, etc.) follow-up audits for all AOs will be conducted at least every three years, or more frequently as determined by DCP.
- Expected ‘for-cause’ on-site auditing may be conducted on an ad hoc basis.
Outline plans for inspecting and auditing CP-CTNet Sites, including, but not limited to, the following activities:
- Develop and/or update as well as implement processes, procedures and documentation for clinical trials auditing of all CP-CTNet Sites;
- On-site or remote auditing of all CTNet Sites to ensure data quality and site compliance with federal regulations, GCP Guidelines, NCI and NIH policies and procedures, and DCP's protocol, regulatory, pharmacy, and data collection requirements (additional information can be found at http://prevention.cancer.gov/cp-ctnet );
- Preparation and distribution of reports of audit visit findings and follow-up of visit action items for NCI DCP review and approval and dissemination to the appropriate LAOs.
- Engage with CP-CTNet Sites and NCI staff to identify areas for systemic and policy-level improvements in order to increase both efficiency and compliance, ensure the protection of human subjects, and enhance the quality and integrity of CP-CTNet clinical trials.
Subsection D: Statistical Support Unit:
Describe proposed approach and plans for providing statistical support for clinical trials data collected within CP-CTNet. Specifically, address support activities for the following:
- Providing independent and authoritative statistical consultations to CP-CTNet constituents for network clinical trials
- Overview of plans to serve the function as the primary statistician(s) of record for all cross-network clinical trials in support of LAOs and AOs in CP-CTNet
Subsection E: Administrative and Coordinating Unit:
Explain how this unit will provide support for administrative and logistical coordination across CP-CTNet operations. Describe plans that include, but are not limited to, the following activities:
- Coordination and standardization of CP-CTNet operational activities across all CTNet Sites, by establishing and maintaining a unified and coordinated operational structure, including processes and documentation that appropriately support and integrate the logistical and administrative requirements of CP-CTNet.
- Development and maintenance of documents, such as the DMASC Manual of Operations, CP-CTNet standard operating procedures, website content for CP-CTNet, and other procedural documents and materials.
- Logistical and administrative support for leadership conference calls every two months, ad hoc conference calls, Steering Committee meetings, annual in-person meeting , and other meetings as required.
- Development, presentation, and dissemination of educational and training content and tools to support clinical and CP-CTNet operations at all CTNet Sites.
- Tools, training, educational materials and other capacity building resources to support timely recruitment to clinical trials at all CP-CTNet Sites.
- Support for the CP-CTNet DSMB meetings, including honoraria for DSMB members (n=5) for yearly remote meetings
- Other operational and administrative support, as needed.
Subsection F: Transition:
Describe a transition plan that will ensure the continuity of all services outlined in this NOFO subsections A-E. The transition plan shall demonstrate a seamless approach of all tasks, systems, URLs, material, data, and other documents securely.
(Additional information can be found at http://prevention.cancer.gov/cp-ctnet .)
Resource Sharing Plan:
Individuals are required to comply with the instructions for the Resource Sharing Plans as provided in the How to Apply - Application Guide .
Other Plan(s):
All instructions in the How to Apply - Application Guide must be followed, with the following additional instructions:
- All applicants planning research (funded or conducted in whole or in part by NIH) that results in the generation of scientific data are required to comply with the instructions for the Data Management and Sharing Plan. All applications, regardless of the amount of direct costs requested for any one year, must address a Data Management and Sharing Plan.
Only limited Appendix materials are allowed. Follow all instructions for the Appendix as described in the How to Apply - Application Guide .
When involving human subjects research, clinical research, and/or NIH-defined clinical trials (and when applicable, clinical trials research experience) follow all instructions for the PHS Human Subjects and Clinical Trials Information form in the How to Apply - Application Guide , with the following additional instructions:
If you answered “Yes” to the question “Are Human Subjects Involved?” on the R&R Other Project Information form, you must include at least one human subjects study record using the Study Record: PHS Human Subjects and Clinical Trials Information form or Delayed Onset Study record.
Study Record: PHS Human Subjects and Clinical Trials Information
Delayed Onset Study
Note: Delayed onset does NOT apply to a study that can be described but will not start immediately (i.e., delayed start). All instructions in the How to Apply - Application Guide must be followed.
Applications must add and complete the Delayed Onset Study record and must check the box "Anticipated Clinical Trial?"
Study Title --use: "Multiple Delayed Onset Studies"
Justification Attachment: Indicate that the clinical trials will be designed and conducted by the CP-CTNet Sites with NCI assistance during the Project Period. Each clinical trial protocol developed will be subject to approval through the standard NCI procedure that involves an initial concept submission and subsequent review. If the concept receives approval, the next stage will be development of the full clinical trial protocol, which will be subject to review and approval by NCI prior to activation through the CP-CTNet. The DMASC will participate as a CP-CTNet clinical trial infrastructure during the implementation of the approved clinical trials.
3. Unique Entity Identifier and System for Award Management (SAM)
See Part 2. Section III.1 for information regarding the requirement for obtaining a unique entity identifier and for completing and maintaining active registrations in System for Award Management (SAM), NATO Commercial and Government Entity (NCAGE) Code (if applicable), eRA Commons, and Grants.gov
Part I. contains information about Key Dates and times. Applicants are encouraged to submit applications before the due date to ensure they have time to make any application corrections that might be necessary for successful submission. When a submission date falls on a weekend or Federal holiday , the application deadline is automatically extended to the next business day.
Organizations must submit applications to Grants.gov (the online portal to find and apply for grants across all Federal agencies). Applicants must then complete the submission process by tracking the status of the application in the eRA Commons , NIH’s electronic system for grants administration. NIH and Grants.gov systems check the application against many of the application instructions upon submission. Errors must be corrected and a changed/corrected application must be submitted to Grants.gov on or before the application due date and time. If a Changed/Corrected application is submitted after the deadline, the application will be considered late. Applications that miss the due date and time are subjected to the NIH Grants Policy Statement Section 2.3.9.2 Electronically Submitted Applications .
Applicants are responsible for viewing their application before the due date in the eRA Commons to ensure accurate and successful submission.
Information on the submission process and a definition of on-time submission are provided in the How to Apply – Application Guide .
5. Intergovernmental Review (E.O. 12372)
This initiative is not subject to intergovernmental review.
All NIH awards are subject to the terms and conditions, cost principles, and other considerations described in the NIH Grants Policy Statement .
Pre-award costs are allowable only as described in the NIH Grants Policy Statement Section 7.9.1 Selected Items of Cost .
Applications must be submitted electronically following the instructions described in the How to Apply - Application Guide . Paper applications will not be accepted.
Applicants must complete all required registrations before the application due date. Section III. Eligibility Information contains information about registration.
For assistance with your electronic application or for more information on the electronic submission process, visit How to Apply – Application Guide . If you encounter a system issue beyond your control that threatens your ability to complete the submission process on-time, you must follow the Dealing with System Issues guidance. For assistance with application submission, contact the Application Submission Contacts in Section VII .
Important reminders:
All PD(s)/PI(s) must include their eRA Commons ID in the Credential field of the Senior/Key Person Profile form . Failure to register in the Commons and to include a valid PD/PI Commons ID in the credential field will prevent the successful submission of an electronic application to NIH. See Section III of this NOFO for information on registration requirements.
The applicant organization must ensure that the unique entity identifier provided on the application is the same identifier used in the organization’s profile in the eRA Commons and for the System for Award Management. Additional information may be found in the How to Apply - Application Guide .
See more tips for avoiding common errors.
Upon receipt, applications will be evaluated for completeness and compliance with application instructions by the Center for Scientific Review and responsiveness by NCI, NIH. Applications that are incomplete, non-compliant and/or nonresponsive will not be reviewed.
Recipients or subrecipients must submit any information related to violations of federal criminal law involving fraud, bribery, or gratuity violations potentially affecting the federal award. See Mandatory Disclosures, 2 CFR 200.113 and NIH Grants Policy Statement Section 4.1.35 .
Send written disclosures to the NIH Chief Grants Management Officer listed on the Notice of Award for the IC that funded the award and to the HHS Office of Inspector Grant Self Disclosure Program at [email protected] .
Applicants are required to follow the instructions for post-submission materials, as described in the policy
Any instructions provided here are in addition to the instructions in the policy.
Section V. Application Review Information
1. criteria.
Only the review criteria described below will be considered in the review process. Applications submitted to the NIH in support of the NIH mission are evaluated for scientific and technical merit through the NIH peer review system.
For this particular announcement, note the following:
A proposed Clinical Trial application may include study design, methods, and intervention that are not by themselves innovative but address important questions or unmet needs. Additionally, the results of the clinical trial may indicate that further clinical development of the intervention is unwarranted or lead to new avenues of scientific investigation.
Overall Impact
Reviewers will provide an overall impact score to reflect their assessment of the likelihood for the project to exert a sustained, powerful influence on the research field(s) involved, in consideration of the following review criteria and additional review criteria (as applicable for the project proposed).
Scored Review Criteria
Reviewers will consider each of the review criteria below in the determination of scientific merit and give a separate score for each. An application does not need to be strong in all categories to be judged likely to have major scientific impact. For example, a project that by its nature is not innovative may be essential to advance a field.
Significance
Does the project address an important problem or a critical barrier to progress in the field? Is the prior research that serves as the key support for the proposed project rigorous? If the aims of the project are achieved, how will scientific knowledge, technical capability, and/or clinical practice be improved? How will successful completion of the aims change the concepts, methods, technologies, treatments, services, or preventative interventions that drive this field?
Are the scientific rationale and need for a clinical trial to test the proposed hypothesis or intervention well supported by preliminary data, clinical and/or preclinical studies, or information in the literature or knowledge of biological mechanisms? For trials focusing on clinical or public health endpoints, is this clinical trial necessary for testing the safety, efficacy or effectiveness of an intervention that could lead to a change in clinical practice, community behaviors or health care policy? For trials focusing on mechanistic, behavioral, physiological, biochemical, or other biomedical endpoints, is this trial needed to advance scientific understanding?
Specific for this NOFO : How likely is the proposed CP-CTNet DMASC to contribute meaningfully to the CP-CTNet goals by enhancing and streamlining data management, data integration and reporting, and clinical trials auditing in the context of multi-site clinical trials research? Will the proposed strategies, infrastructure, and methods adequately support CP-CTNet’s research goals? Do the approaches and methodologies proposed in the application support and enhance the coordination, conduct and quality of cancer prevention clinical trials?
Investigator(s)
Are the PD(s)/PI(s), collaborators, and other researchers well suited to the project? If Early Stage Investigators or those in the early stages of independent careers, do they have appropriate experience and training? If established, have they demonstrated an ongoing record of accomplishments that have advanced their field(s)? If the project is collaborative or multi-PD/PI, do the investigators have complementary and integrated expertise; are their leadership approach, governance, and organizational structure appropriate for the project?
With regard to the proposed leadership for the project, do the PD/PI(s) and key personnel have the expertise, experience, and ability to organize, manage and implement the proposed clinical trial and meet milestones and timelines? Do they have appropriate expertise in study coordination, data management and statistics? For a multicenter trial, is the organizational structure appropriate and does the application identify a core of potential center investigators and staffing for a coordinating center?
Specific for this NOFO: How strong is the background of the PD(s)/PI(s) in leading and coordinating collaborative trans-disciplinary clinical trials research? Do the PD(s)/PI(s) demonstrate accomplishments and related experience in multi-institutional clinical trials data management and reporting, clinical trials auditing, accrual support, biospecimen inventory system management, and logistical coordination activities? How well does the organizational structure support the administrative leadership? How well are the other key personnel (e.g., Functional Unit Directors) proposed in the DMASC applicant team suited and qualified to carry out the activities? Do the DMASC key personnel have complementary and integrated expertise and skills? How well will the team be able to contribute unique expertise and perspectives to such joint endeavors?
Does the application challenge and seek to shift current research or clinical practice paradigms by utilizing novel theoretical concepts, approaches or methodologies, instrumentation, or interventions? Are the concepts, approaches or methodologies, instrumentation, or interventions novel to one field of research or novel in a broad sense? Is a refinement, improvement, or new application of theoretical concepts, approaches or methodologies, instrumentation, or interventions proposed?
Does the design/research plan include innovative elements, as appropriate, that enhance its sensitivity, potential for information or potential to advance scientific knowledge or clinical practice?
Specific for this NOFO: Do the proposed approaches to managing and coordinating DMASC activities include innovative elements, as appropriate, to facilitate efficient and effective operations? How well do the proposed plans for data collection, management, and transmission take advantage of the growing availability of electronic records and interoperability?
Are the overall strategy, methodology, and analyses well-reasoned and appropriate to accomplish the specific aims of the project? Have the investigators included plans to address weaknesses in the rigor of prior research that serves as the key support for the proposed project? Have the investigators presented strategies to ensure a robust and unbiased approach, as appropriate for the work proposed? Are potential problems, alternative strategies, and benchmarks for success presented? If the project is in the early stages of development, will the strategy establish feasibility and will particularly risky aspects be managed? Have the investigators presented adequate plans to address relevant biological variables, such as sex, for studies in vertebrate animals or human subjects?
If the project involves human subjects and/or NIH-defined clinical research, are the plans to address 1) the protection of human subjects from research risks, and 2) inclusion (or exclusion) of individuals on the basis of sex/gender, race, and ethnicity, as well as the inclusion or exclusion of individuals of all ages (including children and older adults), justified in terms of the scientific goals and research strategy proposed?
Does the application adequately address the following, if applicable
Study Design
Is the study design justified and appropriate to address primary and secondary outcome variable(s)/endpoints that will be clear, informative and relevant to the hypothesis being tested? Is the scientific rationale/premise of the study based on previously well-designed preclinical and/or clinical research? Given the methods used to assign participants and deliver interventions, is the study design adequately powered to answer the research question(s), test the proposed hypothesis/hypotheses, and provide interpretable results? Is the trial appropriately designed to conduct the research efficiently? Are the study populations (size, gender, age, demographic group), proposed intervention arms/dose, and duration of the trial, appropriate and well justified?
Are potential ethical issues adequately addressed? Is the process for obtaining informed consent or assent appropriate? Is the eligible population available? Are the plans for recruitment outreach, enrollment, retention, handling dropouts, missed visits, and losses to follow-up appropriate to ensure robust data collection? Are the planned recruitment timelines feasible and is the plan to monitor accrual adequate? Has the need for randomization (or not), masking (if appropriate), controls, and inclusion/exclusion criteria been addressed? Are differences addressed, if applicable, in the intervention effect due to sex/gender and race/ethnicity?
Are the plans to standardize, assure quality of, and monitor adherence to, the trial protocol and data collection or distribution guidelines appropriate? Is there a plan to obtain required study agent(s)? Does the application propose to use existing available resources, as applicable?
Data Management and Statistical Analysis
Are planned analyses and statistical approach appropriate for the proposed study design and methods used to assign participants and deliver interventions? Are the procedures for data management and quality control of data adequate at clinical site(s) or at center laboratories, as applicable? Have the methods for standardization of procedures for data management to assess the effect of the intervention and quality control been addressed? Is there a plan to complete data analysis within the proposed period of the award?
Specific for this NOFO:
- How adequate is the proposed plan for centralized data management and reporting, clinical trials auditing, and administrative and logistical coordination for support of multi-institutional research and collaboration within CP-CTNet? How adequate are plans for providing data management support and developing and managing tools, system(s) and processes for NCI's efforts in tracking and improving participant accrual in CP-CTNet clinical trials? To what extent are the proposed leadership and governance structure, staffing, decision-making processes, and interactions among key personnel optimal for supporting the goals of CP-CTNet?
- How adequate are plans for seamless and secure transition of the CP-CTNet Rave URL, databases, and associated information including the AQuIP system currently being hosted and maintained by the current U24 grantee (i.e., code, database, and/or other AQuIP materials)?
- How adequate are the plans for developing web services to exchange data between systems to assist with knowledge transfer and/or portfolio management and to assist in complying with data reporting requirements?
- How adequate are plans for development and maintenance of the virtual biospecimen inventory system and do the plans include an approach for a seamless and secure transfer of the virtual biospecimen data/inventory from the current U24 grantee?
- How adequately do the plans for independent audits of all CP-CTNet Sites as part of the plan for the functional unit focused on Clinical Trials Auditing address the needs outlined in this NOFO?
- How adequate are the plans for providing statistical expertise and consultation for all CP-CTNet trials and serving as the primary statisticians of record for all cross-network clinical trials in support of CP-CTNet LAOs and AOs?
- How adequately are plans and approaches for providing support for administrative and logistical coordination across CP-CTNet operations described, plans for the development and maintenance of network coordination documents, such as the DMASC Manual of Operations, CP-CTNet standard operating procedures, website content for CP-CTNet, and other procedural documents and materials described? How adequate are plans for the development, presentation, and dissemination of educational and training content and tools to support clinical and CP-CTNet operations and clinical trial recruitment at all CP-CTNet Sites described?
- How adequately does the proposed DMASC organization adequately facilitate (a) the application of novel methodologies and analytics related to data harmonization and the creation of integrated datasets; (b) data management in the context of multi-institutional and interdisciplinary studies; (c) cancer prevention agent development science; (d) appropriate and required privacy and confidentiality protections; (e) regulatory compliance and (g) reuse of data? How strong are the applicant's measures to protect data confidentiality, prevent data loss or data security breaches, and overcome other concerns related to data safety and security?
Environment
Will the scientific environment in which the work will be done contribute to the probability of success? Are the institutional support, equipment, and other physical resources available to the investigators adequate for the project proposed? Will the project benefit from unique features of the scientific environment, subject populations, or collaborative arrangements?
If proposed, are the administrative, data coordinating, enrollment and laboratory/testing centers, appropriate for the trial proposed?
Does the application adequately address the capability and ability to conduct the trial at the proposed site(s) or centers? Are the plans to add or drop enrollment centers, as needed, appropriate?
If international site(s) is/are proposed, does the application adequately address the complexity of executing the clinical trial?
If multi-sites/centers, is there evidence of the ability of the individual site or center to: (1) enroll the proposed numbers; (2) adhere to the protocol; (3) collect and transmit data in an accurate and timely fashion; and, (4) operate within the proposed organizational structure?
Additional Review Criteria
As applicable for the project proposed, reviewers will evaluate the following additional items while determining scientific and technical merit, and in providing an overall impact score, but will not give separate scores for these items.
Study Timeline
Is the study timeline described in detail, taking into account start-up activities, the anticipated rate of enrollment, and planned follow-up assessment? Is the projected timeline feasible and well justified? Does the project incorporate efficiencies and utilize existing resources (e.g., CTSAs, practice-based research networks, electronic medical records, administrative database, or patient registries) to increase the efficiency of participant enrollment and data collection, as appropriate?
Are potential challenges and corresponding solutions discussed (e.g., strategies that can be implemented in the event of enrollment shortfalls)?
Protections for Human Subjects
For research that involves human subjects but does not involve one of the categories of research that are exempt under 45 CFR Part 46, the committee will evaluate the justification for involvement of human subjects and the proposed protections from research risk relating to their participation according to the following five review criteria: 1) risk to subjects, 2) adequacy of protection against risks, 3) potential benefits to the subjects and others, 4) importance of the knowledge to be gained, and 5) data and safety monitoring for clinical trials.
For research that involves human subjects and meets the criteria for one or more of the categories of research that are exempt under 45 CFR Part 46, the committee will evaluate: 1) the justification for the exemption, 2) human subjects involvement and characteristics, and 3) sources of materials. For additional information on review of the Human Subjects section, please refer to the Guidelines for the Review of Human Subjects .
Inclusion of Women, Minorities, and Individuals Across the Lifespan
When the proposed project involves human subjects and/or NIH-defined clinical research, the committee will evaluate the proposed plans for the inclusion (or exclusion) of individuals on the basis of sex/gender, race, and ethnicity, as well as the inclusion (or exclusion) of individuals of all ages (including children and older adults) to determine if it is justified in terms of the scientific goals and research strategy proposed. For additional information on review of the Inclusion section, please refer to the Guidelines for the Review of Inclusion in Clinical Research .
Vertebrate Animals
The committee will evaluate the involvement of live vertebrate animals as part of the scientific assessment according to the following three points: (1) a complete description of all proposed procedures including the species, strains, ages, sex, and total numbers of animals to be used; (2) justifications that the species is appropriate for the proposed research and why the research goals cannot be accomplished using an alternative non-animal model; and (3) interventions including analgesia, anesthesia, sedation, palliative care, and humane endpoints that will be used to limit any unavoidable discomfort, distress, pain and injury in the conduct of scientifically valuable research. Methods of euthanasia and justification for selected methods, if NOT consistent with the AVMA Guidelines for the Euthanasia of Animals, is also required but is found in a separate section of the application. For additional information on review of the Vertebrate Animals Section, please refer to the Worksheet for Review of the Vertebrate Animals Section.
Reviewers will assess whether materials or procedures proposed are potentially hazardous to research personnel and/or the environment, and if needed, determine whether adequate protection is proposed.
Resubmissions
Not applicable
As applicable for the project proposed, reviewers will consider each of the following items, but will not give scores for these items, and should not consider them in providing an overall impact score.
Applications from Foreign Organizations
Not Applicable.
Select Agent Research
Reviewers will assess the information provided in this section of the application, including 1) the Select Agent(s) to be used in the proposed research, 2) the registration status of all entities where Select Agent(s) will be used, 3) the procedures that will be used to monitor possession use and transfer of Select Agent(s), and 4) plans for appropriate biosafety, biocontainment, and security of the Select Agent(s).
Resource Sharing Plans
Reviewers will comment on whether the Resource Sharing Plan(s) (i.e., Sharing Model Organisms ) or the rationale for not sharing the resources, is reasonable.
Authentication of Key Biological and/or Chemical Resources:
For projects involving key biological and/or chemical resources, reviewers will comment on the brief plans proposed for identifying and ensuring the validity of those resources.
Budget and Period of Support
Reviewers will consider whether the budget and the requested period of support are fully justified and reasonable in relation to the proposed research.
2. Review and Selection Process Applications will be evaluated for scientific and technical merit by (an) appropriate Scientific Review Group(s) convened by the NCI, in accordance with NIH peer review policies and practices , using the stated review criteria. Assignment to a Scientific Review Group will be shown in the eRA Commons. As part of the scientific peer review, all applications will receive a written critique. Applications may undergo a selection process in which only those applications deemed to have the highest scientific and technical merit (generally the top half of applications under review) will be discussed and assigned an overall impact score. Appeals of initial peer review will not be accepted for applications submitted in response to this NOFO. Applications will be assigned on the basis of established PHS referral guidelines to the appropriate NIH Institute or Center. Applications will compete for available funds with all other recommended applications submitted in response to this NOFO. Following initial peer review, recommended applications will receive a second level of review by the appropriate national Advisory Council or Board. The following will be considered in making funding decisions: Scientific and technical merit of the proposed project as determined by scientific peer review. Availability of funds. Relevance of the proposed project to program priorities. If the application is under consideration for funding, NIH will request "just-in-time" information from the applicant as described in the NIH Grants Policy Statement Section 2.5.1. Just-in-Time Procedures . This request is not a Notice of Award nor should it be construed to be an indicator of possible funding. Prior to making an award, NIH reviews an applicant’s federal award history in SAM.gov to ensure sound business practices. An applicant can review and comment on any information in the Responsibility/Qualification records available in SAM.gov. NIH will consider any comments by the applicant in the Responsibility/Qualification records in SAM.gov to ascertain the applicant’s integrity, business ethics, and performance record of managing Federal awards per 2 CFR Part 200.206 “Federal awarding agency review of risk posed by applicants.” This provision will apply to all NIH grants and cooperative agreements except fellowships. 3. Anticipated Announcement and Award Dates
After the peer review of the application is completed, the PD/PI will be able to access his or her Summary Statement (written critique) via the eRA Commons . Refer to Part 1 for dates for peer review, advisory council review, and earliest start date.
Information regarding the disposition of applications is available in the NIH Grants Policy Statement Section 2.4.4 Disposition of Applications .
Section VI. Award Administration Information
1. award notices.
A Notice of Award (NoA) is the official authorizing document notifying the applicant that an award has been made and that funds may be requested from the designated HHS payment system or office. The NoA is signed by the Grants Management Officer and emailed to the recipient’s business official.
In accepting the award, the recipient agrees that any activities under the award are subject to all provisions currently in effect or implemented during the period of the award, other Department regulations and policies in effect at the time of the award, and applicable statutory provisions.
Recipients must comply with any funding restrictions described in Section IV.6. Funding Restrictions . Any pre-award costs incurred before receipt of the NoA are at the applicant's own risk. For more information on the Notice of Award, please refer to the NIH Grants Policy Statement Section 5. The Notice of Award and NIH Grants & Funding website, see Award Process.
Individual awards are based on the application submitted to, and as approved by, the NIH and are subject to the IC-specific terms and conditions identified in the NoA.
ClinicalTrials.gov: If an award provides for one or more clinical trials. By law (Title VIII, Section 801 of Public Law 110-85), the "responsible party" must register and submit results information for certain “applicable clinical trials” on the ClinicalTrials.gov Protocol Registration and Results System Information Website ( https://register.clinicaltrials.gov ). NIH expects registration and results reporting of all trials whether required under the law or not. For more information, see https://grants.nih.gov/policy/clinical-trials/reporting/index.htm
Institutional Review Board or Independent Ethics Committee Approval: Recipient institutions must ensure that all protocols are reviewed by their IRB or IEC. To help ensure the safety of participants enrolled in NIH-funded studies, the recipient must provide NIH copies of documents related to all major changes in the status of ongoing protocols.
Data and Safety Monitoring Requirements: The NIH policy for data and safety monitoring requires oversight and monitoring of all NIH-conducted or -supported human biomedical and behavioral intervention studies (clinical trials) to ensure the safety of participants and the validity and integrity of the data. Further information concerning these requirements is found at http://grants.nih.gov/grants/policy/hs/data_safety.htm and in the application instructions (SF424 (R&R) and PHS 398).
Investigational New Drug or Investigational Device Exemption Requirements: Consistent with federal regulations, clinical research projects involving the use of investigational therapeutics, vaccines, or other medical interventions (including licensed products and devices for a purpose other than that for which they were licensed) in humans under a research protocol must be performed under a Food and Drug Administration (FDA) investigational new drug (IND) or investigational device exemption (IDE).
2. Administrative and National Policy Requirements
The following Federal wide and HHS-specific policy requirements apply to awards funded through NIH:
- The rules listed at 2 CFR Part 200 , Uniform Administrative Requirements, Cost Principles, and Audit Requirements for Federal Awards.
- All NIH grant and cooperative agreement awards include the NIH Grants Policy Statement as part of the terms and conditions in the Notice of Award (NoA). The NoA includes the requirements of this NOFO. For these terms of award, see the NIH Grants Policy Statement Part II: Terms and Conditions of NIH Grant Awards, Subpart A: General and Part II: Terms and Conditions of NIH Grant Awards, Subpart B: Terms and Conditions for Specific Types of Grants, Recipients, and Activities .
- HHS recognizes that NIH research projects are often limited in scope for many reasons that are nondiscriminatory, such as the principal investigator’s scientific interest, funding limitations, recruitment requirements, and other considerations. Thus, criteria in research protocols that target or exclude certain populations are warranted where nondiscriminatory justifications establish that such criteria are appropriate with respect to the health or safety of the subjects, the scientific study design, or the purpose of the research. For additional guidance regarding how the provisions apply to NIH grant programs, please contact the Scientific/Research Contact that is identified in Section VII under Agency Contacts of this NOFO.
All federal statutes and regulations relevant to federal financial assistance, including those highlighted in NIH Grants Policy Statement Section 4 Public Policy Requirements, Objectives and Other Appropriation Mandates.
Recipients are responsible for ensuring that their activities comply with all applicable federal regulations. NIH may terminate awards under certain circumstances. See 2 CFR Part 200.340 Termination and NIH Grants Policy Statement Section 8.5.2 Remedies for Noncompliance or Enforcement Actions: Suspension, Termination, and Withholding of Support .
The following special terms of award are in addition to, and not in lieu of, otherwise applicable U.S. Office of Management and Budget (OMB) administrative guidelines, U.S. Department of Health and Human Services (HHS) grant administration regulations at 2 CFR Part 200, and other HHS, PHS, and NIH grant administration policies.
The administrative and funding instrument used for this program will be the cooperative agreement, an "assistance" mechanism (rather than an "acquisition" mechanism), in which substantial NIH programmatic involvement with the recipients is anticipated during the performance of the activities. Under the cooperative agreement, the NIH purpose is to support and stimulate the recipients' activities by involvement in and otherwise working jointly with the recipients in a partnership role; it is not to assume direction, prime responsibility, or a dominant role in the activities. Consistent with this concept, the dominant role and prime responsibility resides with the recipients for the project as a whole, although specific tasks and activities may be shared among the recipients and NIH as defined below.
The PD(s)/PI(s) will have the primary responsibility for:
- Recipients will retain custody of and have primary rights to the data and software developed under these awards, subject to Government rights of access consistent with current HHS, PHS, and NIH policies.
- Provide updates at least annually on progress in PEDP implementation.
- Managing the DMASC’s operations and tasks, including but not limited to data management and reporting, clinical trials auditing, statistical support, and administration and coordination of the CP-CTNet.
- Managing a centralized clinical trial data management system to support CP-CTNet trials including coordinating and leveraging, where feasible, NCI’s Common Data Elements.
- Protecting the confidentiality of CP-CTNet clinical trial data and the information shared with CP-CTNet organizations, including, without limitation, unpublished data, protocols, data analysis, and other confidential information received by CP-CTNet personnel.
- Providing routine and ad-hoc reports and data to CP-CTNet and NCI.
- Developing and supporting the end of study data collection and reporting requirements.
- Developing and/or maintaining a clinical trials accrual quality improvement program and accrual tracking system.
- Developing and maintaining a virtual biospecimen inventory system to support tracking the collection and use of the biospecimens and related data.
- Providing expertise in statistical design and analysis for cross-network trials as the primary statistician of record
- Serving as a resource for consultations on statistical design and analysis for all CP-CTNet trials
- Developing and maintaining procedural documents and materials for logistical and administrative support of CP-CTNet.
- Establishing collaborations among the DMASC team, CP-CTNet, and other NCI support organizations.
- Establishing project timelines in coordination with NCI to ensure all required DMASC activities are adequate and developing standard operating procedures in support of CP-CTNet operations and management.
- Participating in the collective management of the CP-CTNet including the internal evaluation of the CP-CTNet program.
- Coordinating with and leveraging, where feasible, technology from related NCI-sponsored informatics initiatives, e.g. the NCI Informatics Technology for Cancer Research ( ITCR ) program, and the NCI Cancer Research Data Commons ( cbiit.cancer.gov/cancerdatacommons ). Additional information regarding this can be found at http://prevention.cancer.gov/cp-ctnet .
NIH staff have substantial programmatic involvement that is above and beyond the normal stewardship role in awards, as described below:
An NCI Program staff member(s) acting as a Project Scientist(s) will have substantial programmatic involvement that is above and beyond the normal stewardship role in awards, as described below. Additional NCI staff members may be designated to have substantial involvement (as Project Scientists). Additionally, an NCI program director acting as Program Official will be responsible for the normal scientific and programmatic stewardship of the award and will be named in the award notice.
The main responsibilities of substantially involved NCI staff members include, but are not limited to, the following activities:
- Ensuring that plans for data management, data reporting, auditing, and coordination of clinical trials proposed are within the research scope of CP-CTNet and relevant to the state-of-the-science, NIH/NCI priorities, resources, and availability of funding;
- Serving as a resource for best practices for data management, data reporting, and clinical trials auditing;
- Overseeing and participating as necessary in clinical trials audits and quality assurance site visits (on-site and remote) and reviewing audit reports;
- Working with CP-CTNet Awardees to collaboratively manage scientific, administrative, and implementation issues associated with their participation in the conduct of clinical trials across CP-CTNet;
- Informing the CP-CTNet awardee PD(s)/PI(s) of scientific opportunities resulting from NCI-supported clinical research programs and facilitating collaborations between the CP-CTNet and other NCI-sponsored programs;
- Facilitating formal aspects of collaborations with outside organizations including review of any memoranda of understanding and data/material transfer agreements for compliance with NIH/NCI and Federal policies;
- Reviewing accrual and overall performance of CP-CTNet clinical trials at CTNet Sites;
- Reviewing compliance with applicable HHS, FDA, OHRP, NIH, and NCI regulations for clinical research involving human research subjects; and
- Monitoring the progress and performance of the key components of the CP-CTNet.
- Sponsoring strategy sessions, when indicated, to discuss specific research initiatives.
- Final review and approval of requests for use of any biospecimens collected per the approved protocol for CP-CTNet trials.
The NCI will have access to all raw data (including imaging data) from clinical trial participants collected and/or generated under this Cooperative Agreement and may periodically review the data. The NCI may also review all records related to recipients performance under the award for appropriate collection, review, and distribution of biospecimens collected in association with CP-CTNet trials.
The NCI reserves the right to reduce the budget or withhold an award in the event of substantial awardee underperformance or other substantial failure to comply with the terms of award.
Areas of Joint Responsibility include:
Steering Committee : A Steering Committee will be the governing body of CP-CTNet that will integrate the efforts of all CP-CTNet awardees and provide oversight of collaborative activities.
The Steering Committee will consist of the following voting members:
- Two representatives from each CP-CTNet awardee (i.e., from CP-CTNet Sites and from DMASC), one of whom must be the PD/PI, who will jointly have one vote for the CP-CTNet award they represent; and
- NCI Project Scientist(s), who will collectively have one vote for NCI.
The NCI Program Official will be a non-voting member of the Steering Committee.
Additional non-voting members may be added to the committee as needed.
The Steering Committee will be chaired by a PD/PI of a CP-CTNet cooperative agreement award and will be elected by the voting members of the Steering Committee.
Key responsibilities of the Steering Committee include:
- Holding quarterly meetings;
- Developing Network operating policies for the implementation by the CP-CTNet awardees;
- Approving the Manual of Operations for the Network;
- Facilitating the process of joint development of appropriate early phase prevention clinical trial protocols by CP-CTNet Sites and NCI (see below);
- Reviewing and approving the studies that will use Rapid Response Restricted Funds;
- Reviewing and approving proposals for mentoring and professional development of young/junior investigators
- Developing agenda and co-organizing with the NCI the annual in-person investigator meeting to be held at NCI ( https://events.cancer.gov/dcp/iscore ); and
- Other programmatic responsibilities to be addressed jointly, as needed, by the CP-CTNet awardees and the NCI staff.
Subcommittees. The Steering Committee may establish subcommittees for specific purposes (e.g., for joint development of clinical trial protocols by CP-CTNet awardees and NCI staff members, see below). The NCI Project Scientist(s) may serve on such subcommittees, as they deem appropriate. Other NCI staff members may also be involved as needed.
Joint Development of Early Phase Prevention Clinical Trial Protocols by CP-CTNet Awardees and NCI. CP-CTNet awardees will be expected to participate as active team members and work closely with the NCI on the development of appropriate clinical trial protocol. These joint activities will include (but will not be limited to) the following aspects:
- General aspects of collaboration on study development and conduct, especially with respect to compliance with federal regulations for clinical trial research, accrual, and participation in activities related to the collective management of the CP-CTNet, as appropriate;
- Development of concepts for new clinical trials, either in response to specific concept solicitations from NCI or as unsolicited concepts developed by the LAOs or AOs.
- Meeting as frequently as needed to assure optimal study performance and to review studies performed under the CP-CTNet awards.
Dispute Resolution:
Any disagreements that may arise in scientific or programmatic matters (within the scope of the award) between recipients and NIH may be brought to Dispute Resolution. A Dispute Resolution Panel composed of three members will be convened: a designee of the Steering Committee chosen without NIH staff voting, one NIH designee, and a third designee with expertise in the relevant area who is chosen by the other two; in the case of individual disagreement, the first member may be chosen by the individual recipient. This special dispute resolution procedure does not alter the recipient's right to appeal an adverse action that is otherwise appealable in accordance with PHS regulation 42 CFR Part 50, Subpart D and HHS regulation 45 CFR Part 16.
3. Data Management and Sharing
Consistent with the 2023 NIH Policy for Data Management and Sharing, when data management and sharing is applicable to the award, recipients will be required to adhere to the Data Management and Sharing requirements as outlined in the NIH Grants Policy Statement . Upon the approval of a Data Management and Sharing Plan, it is required for recipients to implement the plan as described.
4. Reporting
When multiple years are involved, recipients will be required to submit the Research Performance Progress Report (RPPR) annually and financial statements as required in the NIH Grants Policy Statement Section 8.4.1 Reporting. To learn more about post-award monitoring and reporting, see the NIH Grants & Funding website, see Post-Award Monitoring and Reporting .
A final RPPR, invention statement, and the expenditure data portion of the Federal Financial Report are required for closeout of an award, as described in the NIH Grants Policy Statement Section 8.6 Closeout . NIH NOFOs outline intended research goals and objectives. Post award, NIH will review and measure performance based on the details and outcomes that are shared within the RPPR, as described at 2 CFR Part 200.301.
Section VII. Agency Contacts
We encourage inquiries concerning this funding opportunity and welcome the opportunity to answer questions from potential applicants.
eRA Service Desk (Questions regarding ASSIST, eRA Commons, application errors and warnings, documenting system problems that threaten submission by the due date, and post-submission issues)
Finding Help Online: https://www.era.nih.gov/need-help (preferred method of contact) Telephone: 301-402-7469 or 866-504-9552 (Toll Free)
General Grants Information (Questions regarding application instructions, application processes, and NIH grant resources) Email: [email protected] (preferred method of contact) Telephone: 301-480-7075
Grants.gov Customer Support (Questions regarding Grants.gov registration and Workspace) Contact Center Telephone: 800-518-4726 Email: [email protected]
Eva Szabo, MD National Cancer Institute (NCI) Telephone: 240-276-7011 Email: [email protected]
Referral Officer National Cancer Institute (NCI) Telephone: 240-276-6390 Email: [email protected]
Sean Hine National Cancer Institute (NCI) Telephone: 301-276-6291 Email: [email protected]
Section VIII. Other Information
Recently issued trans-NIH policy notices may affect your application submission. A full list of policy notices published by NIH is provided in the NIH Guide for Grants and Contracts . All awards are subject to the terms and conditions, cost principles, and other considerations described in the NIH Grants Policy Statement .
Awards are made under the authorization of Sections 301 and 405 of the Public Health Service Act as amended (42 USC 241 and 284) and under Federal Regulations 42 CFR Part 52 and 2 CFR Part 200.
IMAGES
COMMENTS
CRN Portfolio The NIHR Clinical Research Network (CRN) Portfolio of studies consists of research studies that are eligible for support from the NIHR CRN in England.
These provide a network of research expertise and clinical leadership to deliver research studies on the NIHR CRN Portfolio of studies. Find out more about our performance and key statistics relating to our activity.
The NIHR Clinical Research Network Portfolio is part of the UK Clinical Research Network Portfolio of studies, which comprises the network Portfolios for England, Northern Ireland, Scotland and Wales. NIHR CRN supported studies benefit from the access to the NIHR CRN Study Support Service - a standard national framework for supporting the ...
The UKCRN Portfolio comprises four smaller portfolios of studies belonging to the four nations of the United Kingdom. A second version of the search tool is accessible from the UKCRN Search tool and allows searches within the specific portfolios of these countries. The links to this are shown in Figure 1 below and a separate User Guide for this ...
To access NIHR Clinical Research Network support in England, and be included on the NIHR CRN Portfolio, studies must satisfy the requirements of the Department of Health and Social Care established Eligibility Criteri.
More. STUDY Search. There is now a new Study Search public app, click this notice to be redirected. A site search / find a site app is being developed and will be available by the time this dashboard is archived. If you have any problems please contact [email protected].
NIHR Clinical Research Network (CRN) Portfolio Adoption onto the portfolio has a number of benefits for researchers, such as help in identifying potential research sites, access to patients and the public to carry out ' PPI ' and advice on recruitment strategy at any point during the study. The CRN offers support to researchers via their Study Support Service and likewise via each ...
The database holds records of all research ongoing in the UK that is eligible for the UKCRN Portfolio. In Scotland, studies funded by eligible funders are entered. NHS R&D offices and Scottish Clinical Research Networks are responsible for creating records on the system, however it is the responsibility of Chief Investigators to ensure that their study record is complete and accurate.
The source of research funding is the principal determinant of eligibility for NIHR portfolio adoption: Studies that are funded by the NIHR and / or other areas of central Government and those which are funded by NIHR non-commercial partners, are automatically eligible for consideration for NIHR CRN support provided they meet the definition of ...
The UK Clinical Research Network The UKCRN consists of the National Institute for Health Research Clinical Research Network (NIHR CRN) in England and its equivalents in the devolved nations. Through the UKCRN, researchers can have access to support at the planning, set-up and delivery phases of a study, including advice and support to overcome barriers to successful recruitment.
The NIHR Clinical Research Network (CRN) Portfolio is a grouping of high-quality clinical research studies that have satisfied certain eligibility criteria.
The Network supports the delivery of a portfolio of clinical research studies, including life-sciences industry studies, across all parts of the NHS in England. It does this by providing funds to hospitals and surgeries to invest in clinical research nurses, and other clinical staff. This highly-trained workforce matches patients with ...
The NIHR Portfolio consists of high-quality clinical research studies across a range of over 26 speciality groups that are eligible for consideration for research support from the Clinical Research Network in England. Portfolio adoption can provide management of current studies, facilitate feasibility of future studies and support with staffing.
The Portfolio Maps can be filtered by a keyword, for example you may wish to search for a specific disease or condition relevant to the research study. This field scans keywords from the long title of the study, the inclusion criteria and the exclusion criteria.
The NCRI Group portfolio maps are a visual overview of the UK cancer clinical studies within the NIHR CRN portfolio, which are preparing to open for recruitment ('In set-up') or are actively recruiting patients ('Open'). The maps categorise studies by disease site, research area and Local Clinical Research Network (LCRN). When sharing or adapting these maps, please cite NCRI Portfolio ...
NIHR CRN supported studies benefit from: Access to the Study Support Service - a standard national framework of support to help you Plan, Place and Perform high quality research in England Access to relevant research delivery training, including Good Clinical Practice training ISRCTN registration via the Central Portfolio Management System (CPMS).
The Clinical Research Network West Midlands (CRN WM) Digital Portfolio Maps have been developed to inform Researchers and Healthcare Professionals of the research studies available locally (West Midlands) and nationally.
The network supports delivery of high-quality research within the NHS in England and supports researchers, through provision of staff and resources, with feasibility, site set-up, patient recruitment and study management. Since 2013, over 80% of commercial contract studies running within the UK sat within the UKCRN Portfolio.
The Clinical Research Network (CRN) West of England, hosted at University Hospitals Bristol and Weston NHS Foundation Trust, facilitates research in NHS, Public Health and social care settings, supporting portfolio studies in 31 specialties in sites such as universities, schools, care homes, hospices, prisons and clinical settings.
Find out more about the BSRBR-RA study at The University of Manchester and its part in the NIHR Clinical Research Network Portfolio.
The NIHR CRN maintains data in order to help coordinate a national portfolio of clinical research. The datasets we hold are used to manage core study information, capacity and capability data, study site information, research activity data and research management information.
Quest Diagnostics DGX, in collaboration with the University of Alabama, conducted a study that suggests adherence to guideline-based laboratory testing and treatment of pregnant women for two of ...
It, therefore, relates only to England. 1.2. The NIHR CRN Portfolio of studies consists of research studies that are eligible for support from the NIHR CRN in England. Studies are required to meet these eligibility criteria to be accepted onto the portfolio and throughout the duration of their delivery. 1.3.
Learn about Lilly's pipeline, including information about our investigational molecules and potential indication data.
In addition, the clinical enterprise embraces a wide range of clinical areas and several multidisciplinary centers, including the NCI-designated Markey Cancer Center. UK Chandler Hospital includes the only Level 1 Trauma Center for both adult and pediatric patients and the only Level IV neonatal intensive care unit in Central and Eastern Kentucky.
Eligibility for NIHR Clinical Research Network support is determined on a study-by-study basis, with emphasis on the study rather than the activity of an individual. If you hold a Research Training Award and the specific project which you are working on underwent protocol peer review then no further peer review is required.
Trans-Network Research Activities. The DMASC and CP-CTNet Sites will be expected to work jointly toward the CP-CTNet network goals by: Collaborating on network activities; Working with network collaborators to develop cross-network clinical trials; and; Participating in high priority ancillary studies, including developmental work for new ...